- Record: found
- Abstract: found
- Article: found
RGC-specific ATF4 and/or CHOP deletion rescues glaucomatous neurodegeneration and visual function

Read this article at
Abstract
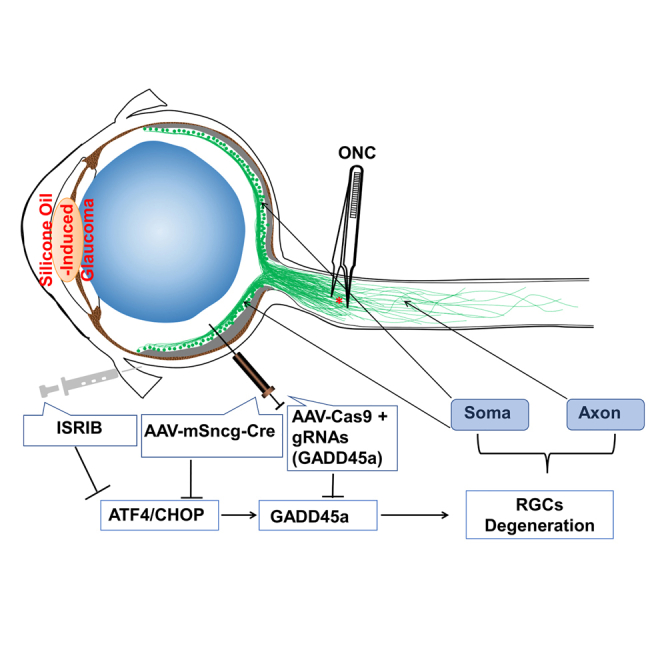
Endoplasmic reticulum (ER) stress has been linked with various acute and chronic neurodegenerative diseases. We previously found that optic nerve (ON) injury and diseases induce neuronal ER stress in retinal ganglion cells (RGCs). We further demonstrated that germline deletion of CHOP preserves the structure and function of both RGC somata and axons in mouse glaucoma models. Here we report that RGC-specific deletion of CHOP and/or its upstream regulator ATF4 synergistically promotes RGC and ON survival and preserves visual function in mouse ON crush and silicone oil-induced ocular hypertension (SOHU) glaucoma models. Consistently, topical application of the ATF4/CHOP chemical inhibitor ISRIB or RGC-specific CRISPR-mediated knockdown of the ATF4 downstream effector Gadd45a also delivers significant neuroprotection in the SOHU glaucoma model. These studies suggest that blocking the neuronal intrinsic ATF4/CHOP axis of ER stress is a promising neuroprotection strategy for neurodegeneration.
Graphical abstract
Abstract
Hu and colleagues present experimental evidence that neuronal intrinsic blocking of ATF4 and/or CHOP, ATF4 downstream molecule, Gadd45a, and local delivery of an ATF4/CHOP inhibitor, ISRIB, prevent glaucomatous neurodegeneration, indicating a promising neuroprotective gene therapy strategy for retinal ganglion cells in traumatic and glaucomatous optic neuropathies.
Related collections
Most cited references76
- Record: found
- Abstract: found
- Article: not found
The unfolded protein response: from stress pathway to homeostatic regulation.
- Record: found
- Abstract: found
- Article: not found
Mechanisms, regulation and functions of the unfolded protein response
- Record: found
- Abstract: found
- Article: not found