- Record: found
- Abstract: found
- Article: found
Management of acquired resistance to EGFR TKI–targeted therapy in advanced non-small cell lung cancer
Read this article at
Abstract
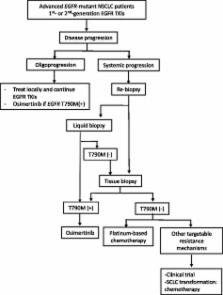
Recent advances in diagnosis and treatment are enabling a more targeted approach to treating lung cancers. Therapy targeting the specific oncogenic driver mutation could inhibit tumor progression and provide a favorable prognosis in clinical practice. Activating mutations of epidermal growth factor receptor ( EGFR) in non-small cell lung cancer (NSCLC) are a favorable predictive factor for EGFR tyrosine kinase inhibitors (TKIs) treatment. For lung cancer patients with EGFR-exon 19 deletions or an exon 21 Leu858Arg mutation, the standard first-line treatment is first-generation (gefitinib, erlotinib), or second-generation (afatinib) TKIs. EGFR TKIs improve response rates, time to progression, and overall survival. Unfortunately, patients with EGFR mutant lung cancer develop disease progression after a median of 10 to 14 months on EGFR TKI. Different mechanisms of acquired resistance to first-generation and second-generation EGFR TKIs have been reported. Optimal treatment for the various mechanisms of acquired resistance is not yet clearly defined, except for the T790M mutation. Repeated tissue biopsy is important to explore resistance mechanisms, but it has limitations and risks. Liquid biopsy is a valid alternative to tissue re-biopsy. Osimertinib has been approved for patients with T790M-positive NSCLC with acquired resistance to EGFR TKI. For other TKI-resistant mechanisms, combination therapy may be considered. In addition, the use of immunotherapy in lung cancer treatment has evolved rapidly. Understanding and clarifying the biology of the resistance mechanisms of EGFR-mutant NSCLC could guide future drug development, leading to more precise therapy and advances in treatment.
Related collections
Most cited references63
- Record: found
- Abstract: found
- Article: not found
Epithelial-mesenchymal transitions in development and disease.
- Record: found
- Abstract: found
- Article: not found
Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors.
- Record: found
- Abstract: found
- Article: not found