- Record: found
- Abstract: found
- Article: found
Locality-sensitive hashing for the edit distance
Read this article at
Abstract
Motivation
Sequence alignment is a central operation in bioinformatics pipeline and, despite many improvements, remains a computationally challenging problem. Locality-sensitive hashing (LSH) is one method used to estimate the likelihood of two sequences to have a proper alignment. Using an LSH, it is possible to separate, with high probability and relatively low computation, the pairs of sequences that do not have high-quality alignment from those that may. Therefore, an LSH reduces the overall computational requirement while not introducing many false negatives (i.e. omitting to report a valid alignment). However, current LSH methods treat sequences as a bag of k-mers and do not take into account the relative ordering of k-mers in sequences. In addition, due to the lack of a practical LSH method for edit distance, in practice, LSH methods for Jaccard similarity or Hamming similarity are used as a proxy.
Results
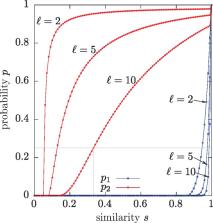
We present an LSH method, called Order Min Hash (OMH), for the edit distance. This method is a refinement of the minHash LSH used to approximate the Jaccard similarity, in that OMH is sensitive not only to the k-mer contents of the sequences but also to the relative order of the k-mers in the sequences. We present theoretical guarantees of the OMH as a gapped LSH.
Availability and implementation
The code to generate the results is available at http://github.com/Kingsford-Group/omhismb2019.
Related collections
Most cited references12
- Record: found
- Abstract: not found
- Article: not found
Binary codes capable of correcting deletions, insertions, and reversals
- Record: found
- Abstract: found
- Article: not found