- Record: found
- Abstract: found
- Article: found
The minimizer Jaccard estimator is biased and inconsistent
Read this article at
Abstract
Motivation
Sketching is now widely used in bioinformatics to reduce data size and increase data processing speed. Sketching approaches entice with improved scalability but also carry the danger of decreased accuracy and added bias. In this article, we investigate the minimizer sketch and its use to estimate the Jaccard similarity between two sequences.
Results
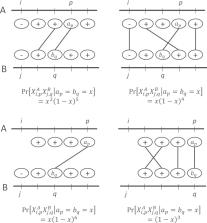
We show that the minimizer Jaccard estimator is biased and inconsistent, which means that the expected difference (i.e. the bias) between the estimator and the true value is not zero, even in the limit as the lengths of the sequences grow. We derive an analytical formula for the bias as a function of how the shared k-mers are laid out along the sequences. We show both theoretically and empirically that there are families of sequences where the bias can be substantial (e.g. the true Jaccard can be more than double the estimate). Finally, we demonstrate that this bias affects the accuracy of the widely used mashmap read mapping tool.
Availability and implementation
Scripts to reproduce our experiments are available at https://github.com/medvedevgroup/minimizer-jaccard-estimator/tree/main/reproduce.
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: not found
Minimap2: pairwise alignment for nucleotide sequences

- Record: found
- Abstract: found
- Article: found
Mash: fast genome and metagenome distance estimation using MinHash

- Record: found
- Abstract: found
- Article: found