- Record: found
- Abstract: found
- Article: found
Gene Selection and Evolutionary Modeling Affect Phylogenomic Inference of Neuropterida Based on Transcriptome Data
Read this article at
Abstract
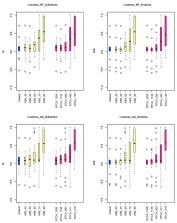
Neuropterida is a super order of Holometabola that consists of the orders Megaloptera (dobsonflies, fishflies, and alderflies), Neuroptera (lacewings) and Raphidioptera (snakeflies). Several proposed higher-level relationships within Neuropterida, such as the relationships between the orders or between the families, have been extensively debated. To further understand the evolutionary history of Neuropterida, we conducted phylogenomic analyses of all 13 published transcriptomes of the neuropterid species, as well as of a new transcriptome of the fishfly species Ctenochauliodes similis of Liu and Yang, 2006 (Megaloptera: Corydalidae: Chauliodinae) that we sequenced. Our phylogenomic data matrix contained 1392 ortholog genes from 22 holometabolan species representing six families from Neuroptera, two families from Raphidioptera, and two families from Megaloptera as the ingroup taxa, and nine orders of Holometabola as outgroups. Phylogenetic reconstruction was performed using both concatenation and coalescent-based approaches under a site-homogeneous model as well as under a site-heterogeneous model. Surprisingly, analyses using the site-homogeneous model strongly supported a paraphyletic Neuroptera, with Coniopterygidae assigned as the sister group of all other Neuropterida. In contrast, analyses using the site-heterogeneous model recovered Neuroptera as monophyletic. The monophyly of Neuroptera was also recovered in concatenation and coalescent-based analyses using genes with stronger phylogenetic signals [i.e., higher average bootstrap support (ABS) values and higher relative tree certainty including all conflicting bipartitions (RTCA) values] under the site-homogeneous model. The present study illustrated how both data selection and model selection influence phylogenomic analyses of large-scale data matrices comprehensively.
Related collections
Most cited references41

- Record: found
- Abstract: found
- Article: found
ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes
- Record: found
- Abstract: found
- Article: not found
Inferring ancient divergences requires genes with strong phylogenetic signals.
- Record: found
- Abstract: found
- Article: not found