- Record: found
- Abstract: found
- Article: found
State-dependent block of Orai3 TM1 and TM3 cysteine mutants: Insights into 2-APB activation
Read this article at
Abstract
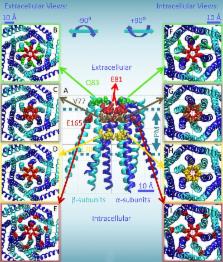
Residue E165, in transmembrane helix 3, participates in formation of the dilated pore of the 2-APB–activated Orai3 channel but not that of the more selective store-operated Orai3 pore.
Abstract
After endoplasmic reticulum (ER) Ca 2+ store depletion, Orai channels in the plasma membrane (PM) are activated directly by ER-resident stromal interacting molecule (STIM) proteins to form the Ca 2+-selective Ca 2+ release-activated Ca 2+ (CRAC) channel. Of the three human Orai channel homologues, only Orai3 can be activated by high concentrations (>50 µM) of 2-aminoethyl diphenylborinate (2-APB). 2-APB activation of Orai3 occurs without STIM1–Orai3 interaction or store depletion, and results in a cationic, nonselective current characterized by biphasic inward and outward rectification. Here we use cysteine scanning mutagenesis, thiol-reactive reagents, and patch-clamp analysis to define the residues that assist in formation of the 2-APB–activated Orai3 pore. Mutating transmembrane (TM) 1 residues Q83, V77, and L70 to cysteine results in potentiated block by cadmium ions (Cd 2+). TM1 mutants E81C, G73A, G73C, and R66C form channels that are not sensitive to 2-APB activation. We also find that Orai3 mutant V77C is sensitive to block by 2-aminoethyl methanethiosulfonate (MTSEA), but not 2-(trimethylammonium)ethyl methanethiosulfonate (MTSET). Block induced by reaction with MTSEA is state dependent, as it occurs only when Orai3-V77C channels are opened by either 2-APB or by cotransfection with STIM1 and concurrent passive store depletion. We also analyzed TM3 residue E165. Mutation E165A in Orai3 results in diminished 2-APB–activated currents. However, it has little effect on store-operated current density. Furthermore, mutation E165C results in Cd 2+-induced block that is state dependent: Cd 2+ only blocks 2-APB–activated, not store-operated, mutant channels. Our data suggest that the dilated pore of 2-APB–activated Orai3 is lined by TM1 residues, but also allows for TM3 E165 to approach the central axis of the channel that forms the conducting pathway, or pore.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function.
- Record: found
- Abstract: found
- Article: not found
STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx.
- Record: found
- Abstract: found
- Article: not found