- Record: found
- Abstract: found
- Article: found
Globular Adiponectin Causes Tolerance to LPS-Induced TNF-α Expression via Autophagy Induction in RAW 264.7 Macrophages: Involvement of SIRT1/FoxO3A Axis
Read this article at
Abstract
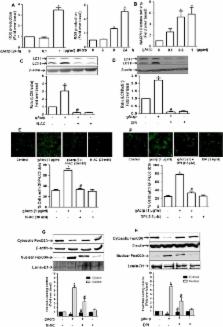
Adiponectin, an adipokine predominantly produced from adipose tissue, exhibited potent anti-inflammatory properties. In particular, it inhibits production of pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α), in macrophages. Autophagy, an intracellular self-digestion process, has been recently shown to regulate inflammatory responses. In the present study, we investigated the role of autophagy induction in the suppression of Lipopolysaccharide (LPS) -induced TNF-α expression by globular adiponectin (gAcrp) and its potential mechanisms. Herein, we found that gAcrp treatment increased expression of genes related with autophagy, including Atg5 and microtubule-associated protein light chain (LC3B), induced autophagosome formation and autophagy flux in RAW 264.7 macrophages. Similar results were observed in primary macrophages isolated peritoneum of mice. Interestingly, inhibition of autophagy by pretreatment with Bafilomycin A1 or knocking down of LC3B gene restored suppression of TNF-α expression, tumor necrosis factor receptor- associated factor 6 (TRAF6) expression and p38MAPK phosphorylation by gAcrp, implying a critical role of autophagy induction in the development of tolerance to LPS-induced TNF-α expression by gAcrp. We also found that knocking-down of FoxO3A, a forkhead box O member of transcription factor, blocked gAcrp-induced expression of LC3II and Atg5. Moreover, gene silencing of Silent information regulator 1 (SIRT1) blocked both gAcrp-induced nuclear translocation of FoxO3A and LC3II expression. Finally, pretreatment with ROS inhibitors, prevented gAcrp-induced SIRT1 expression and further generated inhibitory effects on gAcrp-induced autophagy, indicating a role of ROS production in gAcrp-induced SIRT1 expression and subsequent autophagy induction. Taken together, these findings indicate that globular adiponectin suppresses LPS-induced TNF-α expression, at least in part, via autophagy activation. Furthermore, SIRT1-FoxO3A axis plays a crucial role in gAcrp-induced autophagy in macrophages.
Related collections
Most cited references53
- Record: found
- Abstract: found
- Article: not found
Decoding cell death signals in liver inflammation.

- Record: found
- Abstract: found
- Article: not found