- Record: found
- Abstract: found
- Article: not found
Deriving a Polarizable Force Field for Biomolecular Building Blocks with Minimal Empirical Calibration

Read this article at

Abstract
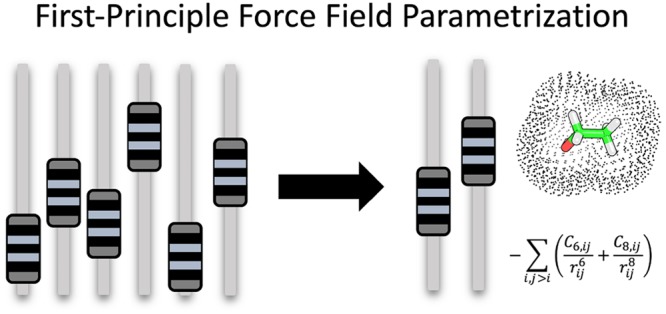
Force field parametrization involves a complex set of linked optimization problems, with the goal of describing complex molecular interactions by using simple classical potential-energy functions that model Coulomb interactions, dispersion, and exchange repulsion. These functions comprise a set of atomic (and molecular) parameters and together with the bonded terms they constitute the molecular mechanics force field. Traditionally, many of these parameters have been fitted in a calibration approach in which experimental measures for thermodynamic and other relevant properties of small-molecule compounds are used for fitting and validation. As these approaches are laborious and time-consuming and represent an underdetermined optimization problem, we study methods to fit and derive an increasing number of parameters directly from electronic structure calculations, in order to greatly reduce possible parameter space for the remaining free parameters. In the current work we investigate a polarizable model with a higher order dispersion term for use in biomolecular simulation. Results for 49 biochemically relevant molecules are presented including updated parameters for hydrocarbon side chains. We show that our recently presented set of QM/MM derived atomic polarizabilities can be used in direct conjunction with partial charges and a higher order dispersion model that are quantum-mechanically determined, to freeze nearly all (i.e., 132 out of 138) nonbonded parameters to their quantum determined values.
Related collections
Most cited references42
- Record: found
- Abstract: found
- Article: not found
Optimized Slater-type basis sets for the elements 1-118.
- Record: found
- Abstract: found
- Article: not found
Water dispersion interactions strongly influence simulated structural properties of disordered protein states.
- Record: found
- Abstract: found
- Article: not found