- Record: found
- Abstract: found
- Article: not found
Convergence of Amyloid-β and Tau Pathologies on Mitochondria In Vivo
Read this article at

Abstract
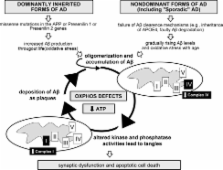
The histopathological characteristics of Alzheimer’s disease (AD) are amyloid-β (Aβ) containing plaques and neurofibrillary tangles (NFTs) as well as neuronal and synaptic loss. Until today, the underlying mechanisms of the interplay of plaques and tangles remained unresolved. There is increasing evidence that mitochondrial dysfunction might be a possible link, as revealed by studies in several APP and tau transgenic mouse models. Recently, we examined mitochondrial function in a novel triple transgenic mouse model (pR5/APP/PS2)— tripleAD mice—that combines both pathologic features of the disease in brain. Using comparative, quantitative proteomics (iTRAQ) and mass spectroscopy, we found a massive deregulation of 24 proteins, of which one third were mitochondrial proteins mainly related to complexes I and IV of the oxidative phosphorylation system (OXPHOS). Remarkably, deregulation of complex I was related to tau, whereas deregulation of complex IV was Aβ dependent, both at the protein and activity levels. The tripleAD mice showed synergistic effects of Aβ and tau already at the age of 8 months, resulting in a depolarized mitochondrial membrane potential. At 12 months, the strongest defects on OXPHOS, synthesis of ATP and reactive oxygen species, were exhibited in the tripleAD mice, again emphasizing synergistic, age-associated effects of Aβ and tau in impairing mitochondria. This review highlights the convergence of Aβ and tau on mitochondria and establishes a molecular link in AD pathology in vivo.
Related collections
Most cited references47
- Record: found
- Abstract: found
- Article: not found
Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice.
- Record: found
- Abstract: found
- Article: not found
High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation.
- Record: found
- Abstract: found
- Article: not found