- Record: found
- Abstract: found
- Article: found
High detection rate for disease-causing variants in a cohort of 30 Iranian pediatric steroid resistant nephrotic syndrome cases
Read this article at
Abstract
Background
Steroid resistant nephrotic syndrome (SRNS) represents a significant renal disease burden in childhood and adolescence. In contrast to steroid sensitive nephrotic syndrome (SSNS), renal outcomes are significantly poorer in SRNS. Over the past decade, extensive genetic heterogeneity has become evident while disease-causing variants are still only identified in 30% of cases in previously reported studies with proportion and type of variants identified differing depending on the age of onset and ethnical background of probands. A genetic diagnosis however can have implications regarding clinical management, including kidney transplantation, extrarenal disease manifestations, and, in some cases, even causal therapy. Genetic diagnostics therefore play an important role for the clinical care of SRNS affected individuals.
Methodology and results
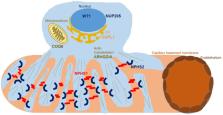
Here, we performed NPHS2 Sanger sequencing and subsequent exome sequencing in 30 consanguineous Iranian families with a child affected by SRNS with a mean age of onset of 16 months. We identified disease-causing variants and one variant of uncertain significance in 22 families (73%), including variants in NPHS1 (30%), followed by NPHS2 (20%), WT1 ( 7%) as well as in NUP205, COQ6, ARHGDIA, SGPL1, and NPHP1 in single cases. Eight of these variants have not previously been reported as disease-causing, including four NPHS1 variants and one variant in NPHS2, ARHGDIA, SGPL1, and NPHP1 each.
Conclusion
In line with previous studies in non-Iranian subjects, we most frequently identified disease-causing variants in NPHS1 and NPHS2. While Sanger sequencing of NPHS2 can be considered as first diagnostic step in non-congenital cases, the genetic heterogeneity underlying SRNS renders next-generation sequencing based diagnostics as the most efficient genetic screening method. In accordance with the mainly autosomal recessive inheritance pattern, diagnostic yield can be significantly higher in consanguineous than in outbred populations.
Related collections
Most cited references43
- Record: found
- Abstract: found
- Article: not found
Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion
- Record: found
- Abstract: found
- Article: found
A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome.
- Record: found
- Abstract: found
- Article: not found