- Record: found
- Abstract: found
- Article: found
DNA methylation dynamics and dysregulation delineated by high-throughput profiling in the mouse
Read this article at
SUMMARY
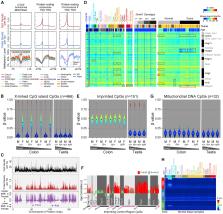
We have developed a mouse DNA methylation array that contains 296,070 probes representing the diversity of mouse DNA methylation biology. We present a mouse methylation atlas as a rich reference resource of 1,239 DNA samples encompassing distinct tissues, strains, ages, sexes, and pathologies. We describe applications for comparative epigenomics, genomic imprinting, epigenetic inhibitors, patient-derived xenograft assessment, backcross tracing, and epigenetic clocks. We dissect DNA methylation processes associated with differentiation, aging, and tumorigenesis. Notably, we find that tissue-specific methylation signatures localize to binding sites for transcription factors controlling the corresponding tissue development. Age-associated hypermethylation is enriched at regions of Polycomb repression, while hypomethylation is enhanced at regions bound by cohesin complex members. Apc Min/+ polyp-associated hypermethylation affects enhancers regulating intestinal differentiation, while hypomethylation targets AP-1 binding sites. This Infinium Mouse Methylation BeadChip (version MM285) is widely accessible to the research community and will accelerate high-sample-throughput studies in this important model organism.
Graphical Abstract

In brief
Infinium arrays are a cost-effective high-throughput DNA methylation profiling tool for human samples. Zhou et al. have developed an equivalent array for the mouse and used it to generate a DNA methylome atlas representing the diversity of DNA methylation biology in development, aging, and disease in this important model organism.
Related collections
Most cited references191
- Record: found
- Abstract: found
- Article: not found
clusterProfiler: an R package for comparing biological themes among gene clusters.

- Record: found
- Abstract: found
- Article: found
BEDTools: a flexible suite of utilities for comparing genomic features
- Record: found
- Abstract: not found
- Article: not found