- Record: found
- Abstract: found
- Article: found
The Trigger Factor Chaperone Encapsulates and Stabilizes Partial Folds of Substrate Proteins
Read this article at
Abstract
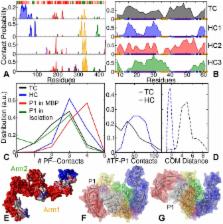
How chaperones interact with protein chains to assist in their folding is a central open question in biology. Obtaining atomistic insight is challenging in particular, given the transient nature of the chaperone-substrate complexes and the large system sizes. Recent single-molecule experiments have shown that the chaperone Trigger Factor (TF) not only binds unfolded protein chains, but can also guide protein chains to their native state by interacting with partially folded structures. Here, we used all-atom MD simulations to provide atomistic insights into how Trigger Factor achieves this chaperone function. Our results indicate a crucial role for the tips of the finger-like appendages of TF in the early interactions with both unfolded chains and partially folded structures. Unfolded chains are kinetically trapped when bound to TF, which suppresses the formation of transient, non-native end-to-end contacts. Mechanical flexibility allows TF to hold partially folded structures with two tips (in a pinching configuration), and to stabilize them by wrapping around its appendages. This encapsulation mechanism is distinct from that of chaperones such as GroEL, and allows folded structures of diverse size and composition to be protected from aggregation and misfolding interactions. The results suggest that an ATP cycle is not required to enable both encapsulation and liberation.
Author Summary
Trigger Factor (TF) is an ATP-independent chaperone protein that assists in folding and prevents misfolding. Up to now, it is a general unsolved question how chaperones assist in the folding of protein chains. Experimental methods that can probe at the length and timescales of inter-residue interactions are scarce, while the systems are too large—and the folding process too long—to be studied by computer simulations. To overcome these obstacles, the authors performed molecular dynamics simulations at key moments along the folding pathway, and address the changes in the folding and unfolding dynamics of protein chains while in contact with TF. This study provides the first detailed view on a chaperone-protein complex in different stages of folding and offers an explanation for the ability of TF to guide chains to their native state. Moreover, the results demonstrates the role of TF’s flexibility in interacting with a wide range of client states. Overall, it explains how TF can interact with many types of substrates in various stages of folding, without the need for an ATP cycle to switch between encapsulation and liberation of client proteins.
Related collections
Most cited references28
- Record: found
- Abstract: found
- Article: not found
Molecular chaperones in protein folding and proteostasis.
- Record: found
- Abstract: not found
- Article: not found
Principles that govern the folding of protein chains.
- Record: found
- Abstract: found
- Article: not found