- Record: found
- Abstract: found
- Article: found
Design of a multi-signature ensemble classifier predicting neuroblastoma patients' outcome
Read this article at
Abstract
Background
Neuroblastoma is the most common pediatric solid tumor of the sympathetic nervous system. Development of improved predictive tools for patients stratification is a crucial requirement for neuroblastoma therapy. Several studies utilized gene expression-based signatures to stratify neuroblastoma patients and demonstrated a clear advantage of adding genomic analysis to risk assessment. There is little overlapping among signatures and merging their prognostic potential would be advantageous. Here, we describe a new strategy to merge published neuroblastoma related gene signatures into a single, highly accurate, Multi-Signature Ensemble (MuSE)-classifier of neuroblastoma (NB) patients outcome.
Methods
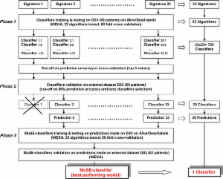
Gene expression profiles of 182 neuroblastoma tumors, subdivided into three independent datasets, were used in the various phases of development and validation of neuroblastoma NB-MuSE-classifier. Thirty three signatures were evaluated for patients' outcome prediction using 22 classification algorithms each and generating 726 classifiers and prediction results. The best-performing algorithm for each signature was selected, validated on an independent dataset and the 20 signatures performing with an accuracy > = 80% were retained.
Results
We combined the 20 predictions associated to the corresponding signatures through the selection of the best performing algorithm into a single outcome predictor. The best performance was obtained by the Decision Table algorithm that produced the NB-MuSE-classifier characterized by an external validation accuracy of 94%. Kaplan-Meier curves and log-rank test demonstrated that patients with good and poor outcome prediction by the NB-MuSE-classifier have a significantly different survival (p < 0.0001). Survival curves constructed on subgroups of patients divided on the bases of known prognostic marker suggested an excellent stratification of localized and stage 4s tumors but more data are needed to prove this point.
Conclusions
The NB-MuSE-classifier is based on an ensemble approach that merges twenty heterogeneous, neuroblastoma-related gene signatures to blend their discriminating power, rather than numeric values, into a single, highly accurate patients' outcome predictor. The novelty of our approach derives from the way to integrate the gene expression signatures, by optimally associating them with a single paradigm ultimately integrated into a single classifier. This model can be exported to other types of cancer and to diseases for which dedicated databases exist.
Related collections
Most cited references42
- Record: found
- Abstract: not found
- Article: not found
Selection of relevant features and examples in machine learning
- Record: found
- Abstract: found
- Article: not found
Robust biomarker identification for cancer diagnosis with ensemble feature selection methods.
- Record: found
- Abstract: found
- Article: not found