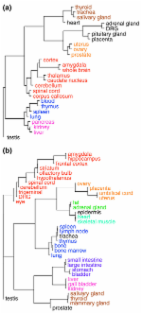
Background The development of an adult from the single cell of a fertilized egg requires a complex orchestration of genes to be expressed at the right time, place, and level. Basic cellular functions require the expression of certain genes in all cells and tissues (that is, in a ubiquitous manner) while specialized functions require restricted expression of other genes in a single or small number of cells and tissues (that is, tissue specific). Both types of genes may be needed for embryonic development as well as for the function of adult cells and tissues. While the details of regulatory mechanisms will vary for individual genes, general features of promoters (and here we will restrict our focus to RNA polymerase II (Pol II) promoters) are likely to facilitate whether a gene will be expressed widely or in a restricted manner. For example, based on the limited number of genes available at the time of the analysis, promoters with CpG islands have been associated with housekeeping genes [1,2]. It is desirable to re-examine this finding in the context of complete genomes for human and mouse and to place it in context with subsequent findings such as the association of CpG islands with embryonic expression [3]. Furthermore, it would also be informative to examine the relationship of CpG islands to the base composition of promoters, and the distribution of motifs thought to be bound by factors closely involved with (or part of) the basal transcription complex. The distribution of major components of the core promoter, the TATA box (TBP/TFIID binding site) and initiator element (Pol II binding site, Inr) [4], and proximal elements such as Yin-Yang 1 (YY1) site [5-8], among genes is not yet well understood. In addition, the functional correlations with tissue specificity and promoter structure are largely unknown beyond the CpG island association. Our goal is to place these components together in general models for tissue specificity using genome-wide surveys of expression in many tissues. Investigators have searched for combinations of transcription-factor-binding sites that confer tissue-specific expression on particular cell types such as muscle [9] or liver [10] in mammals, or in body plan specification in the fruit fly [11,12] (see [13] for a review). In support of these efforts, analyses of genome-wide expression data have largely focused on identifying common patterns for particular tissues, disease states or signaling inputs. For microarray data, investigators have begun defining these patterns, largely through the application of clustering algorithms [14,15]. Our approach is to rank genes in the spectrum of tissue specificity that runs from expression restricted to one tissue to uniform ubiquitous expression. We can study in detail the distribution of human and mouse genes across the spectrum of tissue specificity and use this to identify commonalities and differences in their promoters with the available complete genome sequences [16], libraries enriched for full-length cDNAs [17-19] and genome-wide surveys of gene expression using microarrays [14,20-24], SAGE [25], mRNAs [18] and expressed sequence tags (ESTs) [26]. We validate patterns discovered in human sequence and expression data by comparison to similar mouse data. Measures have been developed for overall tissue specificity [3,27,28] that amount to counting the number of tissues that express a gene. These are really measuring tissue restriction, as they do not consider any bias in the expression levels across the tissues that express the gene. Most specificity measures for a particular tissue are equivalent to the relative expression in a tissue compared to the total expression in all tissues considered, (see, for example [29]). We assert that overall tissue specificity measures should take into account the levels of expression in different tissues, not just presence and absence, and that specificity measures for particular tissues should consider the distribution of expression among all tissues in addition to the tissue of interest. Such measures would enable the correct identification of genes as specific for a tissue when that tissue is not the primary site of expression but there are only a few other tissues where the gene is expressed. A metric for characterizing the breadth and uniformity of the expression pattern of a gene that meets our criteria is the Shannon information theoretic measure entropy. Although entropy has been used previously to identify potential drug targets [30,31] by considering the entropy of the variation of expression levels and to cluster microarray data [32], our direct application of entropy to measuring tissue specificity is unique. Entropy (H) measures the degree of overall tissue specificity of a gene, but does not indicate whether it is specific to a particular tissue. To quantify categorical tissue specificity, we introduce a new statistic (Q) that incorporates overall tissue specificity and relative expression level. We demonstrate that H and Q are effective metrics for ranking and selecting genes according to tissue specificity and then proceed to use them to investigate promoter features (CpG islands, base composition, transcription factor motifs) that may be used distinguish tissue-specific genes from nonspecific genes. The association of promoter features with a quantitative assessment of tissue specificity using H and Q is an important step towards developing models for promoter function. Results Defining tissue specificity We begin by defining the measurement of two kinds of tissue specificity, 'overall' tissue specificity and 'categorical' tissue specificity. (To avoid confusion we will always use the words 'specificity' and 'specific' to refer to the degree of tissue-restricted expression a gene exhibits and never as a synonym for the word 'particular'.) Overall tissue specificity ranks a gene according to the degree to which its expression pattern differs from ubiquitous uniform expression. We use the term 'ubiquitous' expression to mean expression at any level above background in all tissues. Categorical tissue specificity places special emphasis on a particular tissue of interest and ranks a gene according to the degree to which its expression pattern is skewed toward expression in only that particular tissue. In both cases, a gene's specificity to a tissue, cell type or other condition is decreased as the gene is more uniformly expressed in a wider variety of conditions. In addition, the categorical tissue specificity should decrease as the tissue of interest becomes a smaller component of the overall expression pattern of the gene. Given a static multi-tissue expression profile for a gene, there are at least two dimensions along which we can assess the profile to measure tissue specificity. The first dimension is the number of tissues that express the gene above some background level. It can be argued that this dimension measures tissue restriction, that is, a gene shows restricted expression if it is expressed in only a subset of tissues. The second dimension is the uniformity of expression over all tissues that express the gene. A gene that shows significant non-uniform expression is exhibiting tissue-dependent regulation, in addition to any tissue restriction that may be occurring. We assume that a gene that exhibits no tissue-specific regulation will be expressed at the same level in every tissue. We do not assert that such genes are not regulated, only that they are regulated in a way that is not sensitive to tissue. The term 'most tissue-specific' will refer to the range of genes that are closer to the extreme of expression in a single tissue than to the extreme of ubiquitous uniform expression. We will refer to genes close to the uniform and ubiquitous end as either 'least tissue-specific' or 'nonspecific' though the latter term may not be strictly true. The range in the middle will be termed 'semi-tissue specific'. The term 'housekeeping' has been applied to genes that are widely expressed and may show little tissue-specific changes in expression level. We can use such genes as an example of genes that will tend to be ubiquitously and uniformly expressed and thus ought to be nonspecific on average. We will use the phrase 'gene sharing' to refer to the situation that occurs when a gene is tissue-specific, and is expressed in a small number of tissues that can be said to share the gene. Measuring tissue specificity with entropy We used two gene-expression datasets to evaluate our methods; Affymetrix-based data from the GNF Gene Expression Atlas (GNF-GEA) [22] and the distribution of source tissues for EST libraries in the clusters and assemblies of ESTs in the DoTS mouse and human gene index [33]. As described in Materials and methods, the GNF-GEA data were used as provided; EST counts in the DoTS gene index were adjusted with pseudocounts and normalized to account for the different number of ESTs sampled from each tissue across all libraries. Given expression levels of a gene in N tissues, we defined the relative expression of a gene g in a tissue t as p t|g = w g,t /∑1 ≤ t ≤ N w g,t where w g,t is the expression level of the gene in the tissue. The entropy [34] of a gene's expression distribution is H g = ∑1 ≤ t ≤ N - p t|g log2(p t|g ). H g has units of bits and ranges from zero for genes expressed in a single tissue to log2(N) for genes expressed uniformly in all tissues considered. The maximum value of H g depends on the number of tissues considered so we will report this number when appropriate. Because we use relative expression the entropy of a gene is not sensitive to the absolute expression levels. To measure categorical tissue specificity we define Q g|t = H g - log2(p t|g ). The quantity -log2(p t|g ) also has units of bits and has a minimum of zero that occurs when a gene is expressed in a single tissue and grows unboundedly as the relative expression level drops to zero. Thus Q g|t is near its minimum of zero bits when a gene is relatively highly expressed in a small number of tissues including the tissue of interest, and becomes higher as either the number of tissues expressing the gene becomes higher, or as the relative contribution of the tissue to the gene's overall pattern becomes smaller. By itself, the term -log2(p t|g ) is equivalent to p t|g . Adding the entropy term serves to favor genes that are not expressed highly in the tissue of interest, but are expressed only in a small number of other tissues. As described earlier, we want to consider such genes as categorically tissue-specific since their expression pattern is very restricted. Figure 1 shows examples of patterns of GNF-GEA expression data for different values of H g and Q g|t . The top five genes specific to mouse amygdala, lymph node, and liver as assessed by this data are listed in Table 1. Tables of H g and Q g|t values for all genes in all tissues in the GNF-GEA datasets are available in Additional data files 1 and 2. To compare results from microarray and EST-based expression data we mapped the tissues from the GNF-GEA study to the hierarchical controlled vocabulary of anatomical terms used by DoTS and chose a set of 45 tissue terms grouped into 32 groups shown in Table 2. In both cases, the vast majority of genes are widely expressed as measured by H g as shown in Figure 2a. Of the 7,714 probe sets in the GNF-GEA data with an average normalized intensity value above 50 arbitrary units (AU), 6,167 (80%) of genes had H g ≥ 4 bits, which implies expression in at least 16 tissues and typically corresponds to wider, but uneven, expression. Only 87 (2%) of genes had H g ≤ 1.5 bits, which corresponds to expression in as few as three tissues. Both microarray- and EST-based data yielded similar overall curves. The EST curve peaked at a lower H g than the microarray curve. This was due to the small numbers of EST sequences in some of the tissues we considered; EST counts for tissues ranged from 1,933 in the adrenal gland to 331,582 in the central nervous system (CNS). Genes that are ubiquitously expressed may not have ESTs from several of the lightly sequenced tissues, making them appear to have more restricted expression, and hence a lower entropy, than they really do. Figure 2b shows the correlation between estimates of H g derived from microarray and EST data. Visual inspection of the plot reveals that while there are no strong contradictions between the two methods, quantitative agreement is limited. Detailed analysis shows that the standard deviation of the difference of paired H g values is 0.61 bits. Under the null hypothesis that the estimates from the two data sources are totally uncorrelated the average standard deviation was found to be 0.91 bits. We can reject the null hypothesis (P p(G)) in the (-350, -150) region and also a preference for p(A) > p(T) in the -600, -200 region in human (this region is located (-400, -150) in mouse). In tissue-specific CGI+ (Figure 5b) genes the strong C+G bias held but p(C) = p(G), except for the (+50, +100) region where p(C) > p(G). These base-composition differences observed between nonspecific and tissue-specific promoters over regions of hundreds of base-pairs, even in the context of a CpG island, suggest different structural features and regulatory mechanisms for these CGI+ classes. Most striking were differences between nonspecific and tissue-specific promoters that are independent of the presence of a CpG island. A sharp spike in the proportion of A and T was seen in the (-50,-1) region for all classes but was most pronounced in the tissue-specific promoters (Figure 5b,d). These spikes correspond to the presence of a TATA box and suggest a correlation of this motif with tissue-specific genes (explored more fully later). Conversely, all low-specificity genes (Figure 5a,c) shared a common feature in the (+1, +200) region where p(G) > p(C) and p(T) > p(A) that was not seen in tissue-specific genes (Figure 5b,d). As shown later, this low-specificity feature could be partially explained by the presence of a YY1 motif. These base-composition differences observed between nonspecific and tissue-specific promoters are likely to indicate motifs that distinguish the two classes. Selected transcription factor motifs in the core promoter We next examined the distribution of basic core promoter features: the TATA box, the initiator element, and two binding sites for selected ubiquitous transcription factors, Sp1 and YY1, to see if their presence in the proximal promoter was correlated with the tissue specificity of a gene. Two approaches were taken using different datasets and motif-searching methods that gave similar results, providing independent confirmation of results. First, we searched for core motifs using weight matrix hits in promoters of genes selected using H g calculated from the GNF-GEA data. Second, we searched for core motif consensus sites in promoters of genes selected using Q g|t calculated from EST data. TATA boxes are associated with tissue-specific genes We grouped the human genes that expressed at least 200 AU (average value) in the GNF-GEA data by entropy and start CpG island status. The number of genes in each category is shown in Table 5 along with a summary of results. We used alignments of position-specific scoring matrices and scoring thresholds included in the Eukaryotic Promoter Database (EPD) [36] to identify the TATA box and initiator element. Matches to these motifs were preferentially located at the expected positions relative to the transcription start site based on the ratio of the number of observed set to the expected number using a set of random sequences with the same position-dependent base composition as each of the promoters. We searched for the TATA box in the (-45, -10) region where the average observed/expected ratio for the TATA box was 3.1. As shown in Table 5, the most-specific CGI- genes were six times more likely to have a TATA box than the least-specific CGI+ genes (117/215 (54%) versus 183/2072 (9%), P ≈ 0 exact binomial). Similar numbers are found in mouse (52%/11% = 4.7) This trend also holds within CGI- genes and CGI+ genes. The most specific CGI- genes were three times more likely to have a TATA box than the least specific CGI- genes (117/215 versus 110/607, P ≈ 0 exact binomial). While less common in CGI+ genes, TATA boxes were still almost four times as likely to be found in the most specific CGI+ genes than the least specific CGI+ genes (19/56 versus 183/2,072, P = 2 × 10-7 exact binomial). Thus TATA boxes are clearly associated with tissue-specific genes and provide a second axis (with CpG islands) for distinguishing between the most and least specific genes. In contrast, the frequency of occurrences of the initiator element (Pol II binding site) was roughly constant across all tissue-specificity classes for both CGI+ and CGI- genes. We searched for the initiator element in the (-10, +10) region. It occurred in 762 of 1,118 (68%) of CGI- genes and 1,273 of 2,434 (52%) of CGI+ genes. Similarly, it occurred in 149 of 215 (69%) of the most specific genes and 388 of 607 (64%) of CGI+ genes. The observed frequency of TATA+/Inr+ promoters was not significantly different from the expected rate assuming independence of the two individual features (data not shown). Sp1-binding sites are weakly associated with the least tissue-specific genes Sp1 [37,38] is a ubiquitous transcription factor with a G-rich binding site with consensus sequence GGGCGGG that might explain the observed G-richness of the 5' UTR in non-specific genes. We used the GC-box weight matrix and scoring threshold from EPD [36] to identify Sp1 sites. We found that Sp1 sites are preferentially located in the (-150, +1) region in all sets of genes where they occurred on average at twice the expected rate in agreement with previous findings [36]. In both human and mouse, Sp1 sites were rarely found in the 5' UTR despite the G-richness of this region; they occurred at the expected rate of between 2 and 5%. Thus Sp1 sites were not the cause of the G-richness in the 5' UTR. Sp1 sites are associated with CpG islands but are an important component of GGI- promoters as well. Considering just the (-150, +1) region, Sp1 sites occurred in 1,105/2,434 (45%) of human CGI+ gene promoters, and 316/1,118 (28%) of CGI- genes at about 2.5 to 3.0 times the expected frequency in both cases. Frequencies in mouse are 927/2075 (45%) of CGI+ promoters and 464/1652 (28%) CGI- promoters. Sp1 sites were also weakly associated with the least specific genes occurring in 1,105/2,679 (41%) of these genes as compared to 94/271 (32%) in the most tissue-specific genes (P = 0.016). Similar numbers are found in the mouse; 38% of the least specific and 26% of the most specific promoters have Sp1 sites. Thus, although Sp1 shows a preference for the least tissue-specific promoters, it is not a strong predictor of the tissue specificity of a gene. YY1 binding sites are associated with low-specificity genes The transcription factor YY1 [5-8] is also ubiquitously expressed and is thought to bind close to [39] and downstream of the transcription start site. There is evidence that the function of YY1 depends on its orientation [40]. The location and G-richness of the reverse complement consensus sequence (AANATGGCG) make YY1 a candidate for explaining the prominent G > C feature in the (+1, +200) region of low-specificity genes. We consider YY1 because a YY1-like motif was frequently included among the most statistically significant motifs identified by the motif discovery programs AlignACE [41] and MEME [42] in the (+1, +60) region of nonspecific CGI+ promoters (Figure 6a). Our form is most similar to the activating form [43], which may be associated with low-specificity genes. Because of the demonstrated functional sensitivity to the orientation of binding sites we considered each orientation separately. Indeed, as shown in Figure 6b we found each orientation exhibits different position preferences. Sites in the reverse orientation (YY1r) were preferentially located in the (+1, +25) region but with some elevated levels to +80 bp. Start positions of sites in the forward orientation (YY1f) showed a very sharp preference for -3 bp, which probably represents a YY1-like initiator sequence reviewed elsewhere [44]. Both orientations were found predominantly in the least specific genes (Table 5). YY1f initiator sites are rare; only 55/2,679 (2%) were found above background in human low-specificity genes. The rate in mouse, 22/2,832 (0.8%) of low-specificity promoters, is even lower. The YY1r sites are more common and were found above background in 217 (8%) of the 2,679 least specific genes. YY1r sites were more common in CGI+ genes than in CGI- genes (202/2,072 (10%) versus 15/607 (2%) P = 3.7 × 10-9 two-population binomial). The corresponding rates in mouse confirm these observations; 178/2,832 (6%) for all low-specificity genes and 152/1,779 (9%) in CGI+ and 26/1,053 (2%) of CGI- low-specificity promoters. These YY1-like sites therefore constitute a feature strongly associated with the least specific genes and may partially explain the observed G > C ratio in the (+1, +200) region. Q-based analysis of core promoter motifs A second analysis of TATA box and Inr motifs was done to determine if the association of the TATA box with tissue-specific genes is also found in genes ranked by Q and is robust to using EST data as well as promoters that did not specifically rely on full-length cDNA clones. The definition of Q implies that genes with a particular Q-value can have a variety of H g values and thus it may be more difficult to identify features related to tissue specificity. We tabulated all DoTS genes that contained at least two ESTs from an islet-cell library then ranked the genes by Q pancreas computed using EST counts. We used Q pancreas ≤ 7 bits as the criterion for selecting pancreas-specific genes which we grouped into 2-bit Q intervals. For comparison we selected 50 genes with Q pancreas = 8.5 bits, and 50 genes with 10 ≤ Q pancreas ≤ 10.6 bits. Genes with high specificity for the pancreas (0 ≤ Q pancreas ≤ 2 bits, N = 9) preferentially had TATA boxes (8 of 9) with half of these also having an initiation element (4 of 9; Figure 7a). With decreasing specificity, the fraction of genes containing TATA boxes drops with only18 of 81 (2/9) genes with Q > 6 bits having TATA boxes. Thus, the strong correlation of TATA boxes with specific genes found with H g and microarray data was also seen with Q and EST data for pancreas-expressed genes. Also consistent is the observation that initiator elements were found at similar frequencies (around 60%) across all specificity classes (Figure 7b). Similar patterns were observed in other tissues (data not shown). The consistency of findings for the TATA box with human islet genes based on Q and ESTs was next tested with orthologous genes in mouse. This test provides a measure for whether the global pattern observed (TATA box with tissue-specific genes) is also found for the same set of genes in another mammal. We also added bins of genes with higher Q-values that represent more widely expressed genes. For each human gene, the orthologous mouse gene was determined (see Materials and methods for details) and analyzed as described above. Overall, 18.8% of the human genes and 22.9% of the mouse genes that were analyzed carry the TATA box motif. Except for the last group (Q >10 bits) the percentage of the genes with TATA box motifs decreases with the increase in the Q-value. This is to be expected since genes with high Q may be specific to other tissues and hence are more likely to have a TATA box. Discrepancies between human and mouse promoters were noted for only about 10% of all human-mouse pairs analyzed and may reflect sequence differences and possible annotation discrepancies for the transcription start site. Nevertheless, there is overall excellent agreement for the presence of TATA motifs in human and mouse genes. Thus, our assessment of preferential presence of transcription regulatory motifs in the human pancreas-expressed genes also applies to their mouse orthologs. We conclude that genes expressed with restricted tissue-distribution may be preferentially regulated via TATA-mediated transcription, and that genes with broader expression profiles are more likely to be regulated by non-TATA mediated mechanisms (such as YY1). Promoter classes Since the presence or absence of a start CpG island and a TATA box appear to be the primary sequence feature that correlate with tissue specificity, we consider them in more detail. We observe that CpG islands and TATA boxes are not mutually exclusive features of promoters and so we consider all possible combinations of these features. Frequency of promoter classes Figure 8 shows the cumulative fraction of each class of promoter as a function of increasing H g in human (Figure 8a) and mouse (Figure 8b). The data from human and mouse follow similar trends even though the mouse has a lower proportion of CGI+ genes. Overall, CGI+/TATA- genes are the most common, at 50-60% depending on the species. Interestingly, the CGI-/TATA- class is the second most common overall, comprising 20-30% of genes, depending on the species. Genes in this promoter class are roughly equally common across the entire entropy range and are the most common promoters in the mid-specificity range in both species. The classes CGI-/TATA+ and CGI+/TATA+ are the least common (8 to 12% overall). CGI-/TATA+ genes are concentrated in the most specific genes. CGI+/TATA+ are found relatively uniformly across all but the most specific genes. Although the TATA box and CpG islands are strongly predictive of a gene's entropy, Figure 8 also illustrates the limitations of the promoter classes as an explanation for expression patterns. First, although the CGI-/TATA+ and CGI+/TATA- classes are strongly associated with the most and least tissue-specific genes (respectively), instances of genes in each class cover virtually the entire range of tissue specificities. Second, the CGI-/TATA- class is the second most common, illustrating that any degree of tissue specificity can be obtained without these sequence features. Functional assessment of promoter classes using Gene Ontology terms To try to understand the functional correlates of the four promoter classes, we looked for trends in the cellular localization and biological process of the products of genes from each promoter class. We used the DAVID system [45,46], which identifies over-represented Gene Ontology (GO) [47] terms in a set of genes. A summary of the results for human and mouse genes are shown in Table 6. In each case the set of genes in each promoter class were compared to all genes on the corresponding Affymetrix chip. Products of genes in the CGI-/TATA+ class were often (70/198) located extracellularly. Examples of such genes are the insulin-like growth factor family, serum albumin and chymotrypsin. Some extracellular CGI-/TATA+ genes, such as luteinizing hormone beta (LHB) and bone morphogenetic protein 10 (Bmp10) in the mouse, have a high H g because they are not induced in the tissues or at the developmental stages surveyed, but otherwise fit the pattern of secreted proteins. Gene products that are secreted from the cell must be produced at high level to be effective. Indeed we found the maximum expression level of TATA+ genes is higher than TATA- genes; 454/745 (61%) of TATA+ genes express at least 1,000 AU in one or more tissues, whereas only 1,321/3,773 (35%) of TATA- genes express that highly (p-value = 0; two-sample binomial population). A second group of CGI-/TATA+ that is common, but with a p-value just over the p-value cutoff are the muscle contraction-related genes, actin, troponin and members of the myosin family. Products of these genes are also required in large amounts to create the contractile apparatus but are only produced in a few cell types. The biological processes that are enriched in the CGI-/TATA+ class differ between human and mouse, but nearly all of them are descendants of the GO term 'response to stimulus' (GO:0050896). The CGI+/TATA- promoters produce proteins that are typically located in the cell, especially in the cytoplasm and mitochondrion. These locations are consistent with many housekeeping functions. The human results for biological process suggests a large number of housekeeping processes, but these were not confirmed in the mouse using all CGI+/TATA- genes. When we consider just the least specific CGI+/TATA- mouse genes (4.45 ≤ H g ≤ 5.57 bits), we find cellular locations (including the nucleus) and biological processes that match the human results. No significant concentrations of cellular locations or biological processes were found among the CGI+/TATA+ genes. A manual examination of genes in both human and mouse identifies a number of heat-shock proteins, histones and ribosomal proteins although these are not statistically significant as a result of the multiple testing correction. Many of these genes fit the expected expression pattern in that they are widely expressed and at high levels. Interestingly, the products of CGI-/TATA- genes are often located in the plasma membrane (244/499 of human genes with a cellular location) and support signaling and response to the environment. Such products, for example, bradykinin receptor B2, prolactin receptor or protocadherin 9, may be expressed in a tissue-specific pattern, but not at the high levels required for secreted proteins. The exact biological process GO terms that are statistically significant vary between mouse and human, but a common core includes defense response (GO:0006952), immune response (GO:0006955) and response to stimulus (GO:0050896). Thus these genes are similar to CGI-/TATA+ genes in that they are involved in response, but are not (typically) required to be expressed at such high levels. Discussion We have applied Shannon entropy as a novel measure of overall tissue specificity of gene expression and have created a new statistic Q to assess the categorical specificity of a gene for a particular tissue. We have evaluated the performance of entropy on microarray-and EST-based estimates of tissue-specific expression and found that it correctly identifies both tissue-specific and housekeeping genes. Ranking and binning genes by entropy allowed us to begin to deconstruct core promoters into features directing when and where the gene will be expressed. We verified and extended previous observations [2] about the correlation of CpG islands with housekeeping genes and embryonic genes. We then identified differences in the base composition profile of promoters of tissue-specific and nonspecific genes. Next, we identified correlations between, on the one hand, the TATA box and tissue-specific genes, and on the other hand, the YY1 site and nonspecific genes. Finally, we identified trends in promoter classes based on CpG island and TATA box status and associated them with common cellular locations and biological processes. Similar observations were also observed for TATA box and Q-selected genes in pancreas. The identification of an association between promoter type and cellular location and biological function, while an important step in a fundamental understanding of biology, also has practical significance, as the genes in the CGI-/TATA+ and CGI-/TATA- classes are enriched for tissue-specific extracellular and cell surface proteins. Such genes are likely to be useful drug targets. Thus entropy H g and Q have allowed us to discover fundamental properties of mammalian Pol II promoters and should allow serve to aid understanding of expression in particular tissues of interest. The validity of our approach is supported by findings in other work and by the fact that they are robust with respect to the algorithm used to process the expression data. Our finding that most genes are regulated in a tissue-dependent manner is consistent with another analysis of gene expression [14], which found that housekeeping genes cluster in a tissue-specific manner. Thus, it appears, even the most basic biological functions are subject to regulation. The tissue trees we produced contain relationships similar to those in an analysis [48] of mid-specificity genes, including the close relation between lung, and the immune system-related organs spleen and thymus. That analysis is based on a different method and a different set of expression data gives us confidence that Q g|t is properly identifying genes that are specific to a tissue. The GNF-GEA expression data we analyzed was processed with the MAS4 [49] algorithm. We reanalyzed the data from this study after reprocessing it with the more recent Robust Multichip Average (RMA) algorithm [50]. This algorithm tends to suppress low-level signals and we found that most genes appeared to be more tissue specific, that is, had lower H, in the RMA-processed data compared to the reported values. Although this affects some of the precise values of numbers we have reported it does not alter any of the fundamental trends or results. We include tissue specificities based on both analyses in Additional data files 1 and 2. Our analysis focused on only a few sequence features and although we found good correlations, two aspects of our results indicate that there are other regulatory mechanisms not yet identified. First, there is a gradual transition in the frequency of the TATA box and CpG islands between the most and least tissue-specific genes. Second, while these features are strong indicators of high and low specificity, they are far from perfect predictors. Indeed, the middle range of entropies contains a mix of all promoter classes in large numbers, indicating that it is possible to achieve tissue-specific expression with any promoter class. YY1 may be an example of such a supplementary mechanism. While occurring in only 16% of genes, it is very strictly confined to low-specificity genes and is a better indicator of low specificity than CpG islands. We expect that other such signals will be found. Anatomical resolution is an issue with the datasets used in this study. For example, the pancreas consists of exocrine cells, ductal cells and islet cells of several types. The bulk pancreas was used to generate the GNF-GEA data, so the reported expression level is the average mRNA concentrations weighted by the cell-type count. This approximation reduces the maximum possible entropy and, more significantly, can make the apparent entropy different from the true entropy. Genes highly and specifically expressed in a cell type with a small population may currently appear to be ubiquitous with very low overall expression. Genes expressed in a few tissues may be revealed to be less tissue specific as more cell types are measured in detail. Genes that appear to be ubiquitously expressed may turn out to not to be expressed in a few cell types. It will be interesting to see whether data with higher anatomical resolution will help to increase the accuracy of the rules we have identified here for identifying tissue-specific and nonspecific promoters. Our method can be also applied to other sources of expression data including SAGE, reverse transcription PCR (RT-PCR) and in situ hybridization data. SAGE has the advantage of sensitivity, as these studies generally sequence to much greater depths than EST libraries [51]. In situ hybridization data may increase the anatomical resolution of the data. Qualitative intensities, for example, '0', '+', or '+++', can be converted to representative numeric values as appropriate. Our method can also be applied to other collections of conditions beside normal tissues, for example, different types of cancers or samples of the same cancer from multiple patients. Modification of our method to account for temporal changes in tissue specificity represents another direction for future work. The analysis presented here focuses on genes rather than on transcripts generated from different promoters from the same gene. The rate of the occurrence of alternative transcription start sites is at least 9% [52] and may be as high as 25% [53]. The promoters we used were specified by the DBTSS dataset but there may be alternative promoters with different characteristics and tissue-specific usage patterns. Analyses based on different RNA species can easily be incorporated into our approach and is an area of future research. Our results for CpG island frequency in very tissue-specific genes are lower than recent reports [3] that were based upon present/absent calls, that is, tissue counting, using ESTs to measure tissue specificity. This may be due to two reasons. First, as we described in Results, a significant fraction of genes will show no evidence of expression in poorly sampled tissues. A poorly sampled nonspecific gene will appear therefore more tissue specific than it actually is and this increases the number of apparently tissue-specific genes with CpG islands. Second, when we use microarray data and determine tissue specificity by counting tissues expressing above the median value of 200 AU, we see (data not shown) rates of CpG island occurrence in 'specific' genes similar to those reported in [3]. Thus, we conclude that including the variation of expression levels rather than mere presence/absence is important for identifying very tissue-specific genes as assessed by start CpG islands. These results present an initial look at the correlation between tissue specificity, CpG islands and binding sites for selected transcription factors that interact with the basal transcription apparatus. Using a novel approach with entropy-based metrics, we have begun to lay out the framework for promoter function by identifying strong correlations between tissue-specific or ubiquitous expression and a number of these sequence features. We plan to extend this work in several ways. First, we plan to identify correlations with other known transcription-factor-binding sites and novel motifs identified as over-represented in promoter regions [54]. Second, these results will help to understand regulation by combinations of multiple upstream transcription factors in genes specific to particular tissues or clusters of tissues. Conclusions We have used Shannon entropy to quantify and rank the tissue specificity of genes using tissue-survey data. First, this has allowed us to assess the prevalence of tissue-specific regulation; we find that most genes show evidence of some degree of tissue-dependent variation in expression levels. It has also allowed us to find and evaluate associations between promoter features and tissue specificity. We have verified and extended understanding of known associations between, on the one hand, CpG islands and the least tissue-specific genes and, on the other hand, the TATA box and the most tissue-specific genes. However, they are not the sole determinants of tissue-specific expression, as indicated by mid-specificity genes that exhibit a mix of all promoter classes. The class of CGI-/TATA- promoters has emerged as the second most common class of promoter overall and the most common promoter class in mid-specificity genes. Therefore, additional determinants of tissue specificity remain to be found. We have identified one potential determinant, a downstream YY1 site, which is very strongly associated with the least tissue-specific genes but is a relatively rare feature of these promoters. Finally, we have also been able to associate trends in the localization and function of protein products of genes according to their promoter class. Many of the CGI-/TATA+ genes code for highly expressed, very tissue specific, extracellular proteins involved in a cell's response to the environment. CGI-/TATA- genes are also involved in response to the environment, but are found more uniformly across the spectrum of tissue specificity, are not as highly expressed as CGI-/TATA+ genes, and very often code for membrane-bound proteins. CGI+/TATA- genes are more likely to be located in the cytoplasm or nucleus and, as expected, carry out housekeeping functions. All of the results we report are found in both human and mouse and so may reflect general principles of all mammalian species. Materials and methods Processing GNF-GEA [22] and DoTS [33] data The GNF-GEA data are processed as described [22]. Given a set of N tissues we define p t|g = w g,t /∑1 ≤ t ≤ N w g,t where w t is the expression level of the gene g in tissue t. DoTS, available through the AllGenes [33] site, contains ESTs and mRNAs assembled into transcripts that are then clustered into genes. We did not consider any transcript that contains only one EST as this may represent a spurious sequence and did not consider any gene with fewer than five ESTs because they provide a poor estimate of H g . To accommodate the great disparity in sampling depth across tissues we normalized EST counts by tissue. To avoid artificially low entropies for genes that contain relatively few ESTs we used pseudocounts to smooth the data. The expression level of a gene in a tissue is computed as w g,t = (n g,t + 1)/(N t + N g ) where n g,t is the number of ESTs from libraries for a tissue included in a gene, N t is the total number of ESTs from a tissue assembled into genes, and N g is the number of genes. We used different sets of tissues depending on the task. H g and Q measures in Figure 1 used the full GNF-GEA mouse set with a few modifications; adipose tissue and brown fat were merged, epidermis and snout epidermis were merged, digits and tongue were not considered as they are both a combination of skeletal muscle and epidermis. The expression level for a set of merged tissues is the median of the individual tissue replicate medians. For comparison of microarray and EST data we used a set of 27 tissues that were common to both datasets and merged the CNS and peripheral nervous system tissues. Estimating variance To estimate the variance in H and Q, we took advantage of tissue replicates in the GNF-GEA data. Using the mouse dataset, we repeatedly sampled one of the measurements from each pair of replicates and computed H for each gene. We then computed the variance of the distribution of the estimates of H for each gene and show the survivor distribution function in Figure 2. The variance of Q was computed in a similar manner. Clustering tissues Clustering was based on the Q scores for the set of mouse genes with Q g|t ≤ 7 for at least one tissue and expressing at least 200 AU in at least one tissue in the GNF-GEA data. There were 1,786 Affymetrix probe sets selected. The tree in Figure 3 was built by sampling 5,000 sets of 1,000 probe sets and clustering tissues using Pearson correlation and a centered measure using the XCLUSTER [55] program. The consensus tree was built using the program CONSENSE in the PHYLIP [56] package with the default parameters. Identifying genes specific to a set of tissues The total entropy of all tissues under a node can be computed at each node in the hierarchy using a generalization of the grouping theorem [57]. If the entropy of a gene at a node is close to the maximum possible entropy for the number of tissues under the node, then we select it and compute a Q g,n for the gene at the node. Using Q g,n we can rank genes by specificity to a cluster of tissues just as we can for an individual tissue. Predicting CpG islands We predicted CpG islands using the program NEWCGREPORT in the EMBOSS [58] package with the default parameters which require a minimum length of 200 bp, C+G fraction of 0.6 and ratio of observed over expected CpG of 0.5. Statistical significance in embryonic expressed genes We computed statistical significance of differences between all embryonic-expressed genes and adult-specific rates using a hypergeometric distribution. We start with a collection of N CGI+ genes, n e of which are expressed in the embryo, that is, marked as special. The N A tissue-specific genes in the adult are considered a random sample from the original N and we compute the probability of finding that at least (or at most) n ae of these were expressed in the embryo. Modeling distribution of entropy from uniform genes To model the effect of experimental variability, we computed the distribution of the difference between expression levels of individual replicates for each gene and tissue and the mean expression level across replicates as a function of the mean expression level. This distribution was well fit by an exponential distribution with a parameter that depends on the mean expression level. Thus, given an 'ideal' expression level, we can estimate what the experimental variability will be. To model a uniformly expressed gene, we assume that a gene has some average expression level across all tissues and then allow the expression levels in individual tissues to follow a narrow distribution of random fold changes from that level. Specifically, we assumed that the log base 2 of the fold changes is distributed according to a normal distribution with mean equal to 0 and a standard deviation (s). The standard deviation can be adjusted to control the amount of biological variation a 'uniformly' expressed gene is allowed to show. For example, setting s = 0.5 means that about 68% of the fold changes between a particular tissue and the nominal level are within 1.4 up or down from the nominal level, that is, a twofold change from the lowest to the highest levels. Larger fold changes are expected to occur in 32% of tissues. This model allows significant variation and so is arguably close to the upper limit of variation allowable for a gene that shows no tissue specificity. We also used s = 0.25 as a more stringent definition of uniform expression. We sampled mean expression levels from the distribution of observed mean expression levels and sampled entropy values from the probability model. An entropy threshold was estimated by sampling approximately 5,000 random expression profiles and determining the value for a p-value of 0.002. This process was repeated 10 times and the corresponding thresholds and fraction of genes were computed. The thresholds spanned a range of less than 0.01 bit. The tissue-dependent gene fractions never varied by more than one percentage point in either direction. Statistical significance of co-occurrence We estimated the statistical significance of the co-occurrence of motifs using the hypergeometric distribution. Given two motifs with occurrence counts n 1 and n 2, measured in the same set of N promoters, and a co-occurrence count of n 12, we compute the significance as the probability of finding no more than (or at least) n 12 hits in a random selection of n 2 promoters from a pool of N promoters where n 1 of them are 'special'. Comparison of frequency on independent sets Given two sets of size N 1 and N 2 and positive observations n 1 and n 2 in each, we computed the probability that the underlying rates are different using an exact calculation of the binomial distribution to compute the probability of seeing at least (or no more) than n i matches in N i trials where the rate is assumed to be r = n j /N j . We estimated r using the larger of the two sets. Two binomial populations We used the normal approximation to the difference of the proportions normalized by their variance to compute a z-score. Promoter sequences We obtained promoter sequence in two ways. The H-based set of analyses used links from Affymetrix probe sets to RefSeq identifiers to select alignments from the DBTSS promoter sequences covering the (-1000, 200) region downloaded from the DBTSS website [59]. The Q-based analyses of TATA box and initiator elements used genomic locations of DoTS genes on UCSC Golden Path release mm3 [60,61] to identify gene names. Promoter sequences consisting of the 350 bp of the upstream region were then extracted from Ensembl [62]. The mouse homologs were also used as annotated in Ensembl. Core motifs The H-based analysis used core promoter element models from EPD [36,63]. The fraction of promoters containing each matrix was determined as follows for each set of genes (with and without CpG islands in each entropy bin) individually. Having verified that the positional distribution of each motif was sharply peaked at the appropriate place in the promoter sequences ((-40, -20) region for TATA and (-20, +20) region for the initiator element) we considered only the predictions in these windows from all genes. We used the log-likelihood function to score each subsequence against each matrix using the published score cut-offs. The YY1 motif was found in essentially every run of AlignACE and MEME performed on the downstream regions of ubiquitous CGI+ promoters. We explored different motif widths and other settings and selected version that achieved a combination of good coverage and conservation. In all cases we estimated the background rate of random occurrence of motifs by repeatedly scrambling the individual sequences over a 10 bp window to create approximately 1,000 test sequences for each combination of CpG island status and specificity range. These sequences were scored in the same manner as the unscrambled sequences. We estimated the statistical significance of differences of observed frequencies using exact computation of the binomial distribution. The Q-based analyses of core motifs used the TATA box motif (TATAA) and initiator element (YYANWYY). Motif searches were carried out using the tool patternmatch from the biological workbench 3.2 [64]. Only the TATAA instance located closest to the start of the mRNA's alignment to the genome was used. Matches to the initiator element were required to be downstream of the TATAA box when present. YY1 motif We used an AlignACE-derived weight matrix (shown in Figure 6a) to assess the occurrence of YY1-like sites as it contained the YY1 consensus and was built using approximately 100 sites which is many more than previously published weight matrices [43,65] also shown in Figure 6a. GO association analysis We submitted Affymetrix probe set ids of interest to the DAVID website [45,46] and compared them either to all probe sets on the appropriate Affymetrix chips or to all genes in the selected entropy range. We compensated for multiple testing by requiring the reported p-values be better than either 0.05/1472 = 0.00003 (cellular component) or 0.05/8972 = 0.000006 (biological process) using the number of GO terms for the corresponding GO divisions in a Bonferroni correction. RMA quantification We obtained CEL files for the GNF-GEA study from and re-quantified them using the gcrma package [66] in the Bioconductor [67] project for the R statistical analysis program [68]. We use the gcrma options 'type=c('fullmodel')' and 'fast=T'. Additional data files Two additional data files are available with the online version of this article. They contain H and Q values for all normal tissues in the GNF-GEA data set for both human (Additional data file 1) and mouse (Additional data file 2) using both the MAS4 and RMA quantification methods. The RMA data were normalized to yield a common median of 3.75 (human) and 3.22 (mouse) prior to the H and Q calculation. The files are in Excel format. The data for each tissue are placed in separate worksheets. Each worksheet contains H- and Q-values, the expression value of the gene in the worksheet's tissue, and its maximum expression across all tissues in the file, the gene symbol, RefSeq, SwissProt, and Unigene ID, and a description. The rows in each worksheet are sorted by increasing values of Q using the RMA data. Thus the top of each worksheet displays the genes most specific to that worksheet's tissue. Supplementary Material Additional File 1 A table showing H and Q values for all normal human tissues in the GNF-GEA dataset. H and Q values for all normal tissues in the GNF-GEA dataset for human using both the original MAS4 quantification and our RMA re-quantification. The RMA data were normalized to yield common medians of 3.75 prior to the H and Q calculation. The data for each tissue are placed in separate worksheets. Each worksheet contains H- and Q-values, the expression value of the gene in the worksheet's tissue, and its maximum expression across all tissues in the file, the gene symbol, RefSeq, SwissProt, and Unigene ID, and a description. The rows in each worksheet are sorted by increasing values of Q using the RMA data. Thus the top of each worksheet displays the genes most specific to that worksheet's tissue. Click here for file Additional File 2 A table showing H and Q values for all normal mouse tissues in the GNF-GEA dataset. H and Q values for all normal tissues in the GNF-GEA dataset for mouse using both the original MAS4 quantification and our RMA re-quantification. The RMA data were normalized to yield common medians of 3.22 prior to the H and Q calculation. The data for each tissue are placed in separate worksheets. Each worksheet contains H- and Q-values, the expression value of the gene in the worksheet's tissue, and its maximum expression across all tissues in the file, the gene symbol, RefSeq, SwissProt, and Unigene ID, and a description. The rows in each worksheet are sorted by increasing values of Q using the RMA data. Thus the top of each worksheet displays the genes most specific to that worksheet's tissue. Click here for file