- Record: found
- Abstract: found
- Article: found
Natural history study and statistical modeling of disease progression in a preclinical model of myotubular myopathy
Read this article at
ABSTRACT
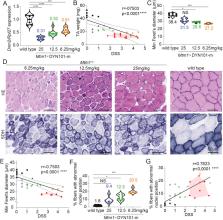
Generating reliable preclinical data in animal models of disease is essential in therapy development. Here, we performed statistical analysis and joint longitudinal–survival modeling of the progressive phenotype observed in Mtm1 −/y mice, a reliable model for myotubular myopathy. Analysis of historical data was used to generate a model for phenotype progression, which was then confirmed with phenotypic data from a new colony of mice derived via in vitro fertilization in an independent animal house, highlighting the reproducibility of disease phenotype in Mtm1 −/y mice. These combined data were used to refine the phenotypic parameters analyzed in these mice and improve the model generated for expected disease progression. The disease progression model was then used to test the therapeutic efficacy of Dnm2 targeting. Dnm2 reduction by antisense oligonucleotides blocked or postponed disease development, and resulted in a significant dose-dependent improvement outside the expected disease progression in untreated Mtm1 −/y mice. This provides an example of optimizing disease analysis and testing therapeutic efficacy in a preclinical model, which can be applied by scientists testing therapeutic approaches using neuromuscular disease models in different laboratories.
This article has an associated First Person interview with the joint first authors of the paper.
Abstract
Summary: This study optimized disease severity analysis and modeled disease progression in Mtm1 −/y mice, and confirmed this model using therapeutic Dnm2 reduction in a dose–response analysis.
Related collections
Most cited references22
- Record: found
- Abstract: found
- Article: not found
A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast.
- Record: found
- Abstract: found
- Article: not found
Mice lacking microRNA 133a develop dynamin 2–dependent centronuclear myopathy.
- Record: found
- Abstract: found
- Article: not found