- Record: found
- Abstract: found
- Article: not found
Reference-based phasing using the Haplotype Reference Consortium panel

Abstract
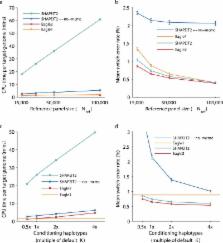
Haplotype phasing is a fundamental problem in medical and population genetics. Phasing is generally performed via statistical phasing within a genotyped cohort, an approach that can attain high accuracy in very large cohorts but attains lower accuracy in smaller cohorts. Here, we instead explore the paradigm of reference-based phasing. We introduce a new phasing algorithm, Eagle2, that attains high accuracy across a broad range of cohort sizes by efficiently leveraging information from large external reference panels (such as the Haplotype Reference Consortium, HRC) using a new data structure based on the positional Burrows-Wheeler transform. We demonstrate that Eagle2 attains a ≈20x speedup and ≈10% increase in accuracy compared to reference-based phasing using SHAPEIT2. On European-ancestry samples, Eagle2 with the HRC panel achieves >2x the accuracy of 1000 Genomes-based phasing. Eagle2 is open source and freely available for HRC-based phasing via the Sanger Imputation Service and the Michigan Imputation Server.
Related collections
Most cited references14
- Record: found
- Abstract: found
- Article: not found
A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase.
- Record: found
- Abstract: found
- Article: not found
Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation.
- Record: found
- Abstract: found
- Article: not found