- Record: found
- Abstract: found
- Article: found
Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity
Read this article at
Significance
Two influenza B viruses (Victoria and Yamagata) cocirculate in humans and contribute to the estimated 290,000–650,000 annual influenza-attributed deaths. Here, we analysed influenza B genomic data to understand the causes of a recent surge in human influenza B infections. We found that evolution is acting differently on Yamagata and Victoria viruses and that this has led to the cocirculation of a diverse group of influenza B viruses. If this phenomenon continues, we could potentially witness the emergence of 3 or more distinct influenza B viruses that could require their own vaccine component, thereby complicating influenza vaccine formulation and highlighting the urgency of developing universal influenza vaccines.
Abstract
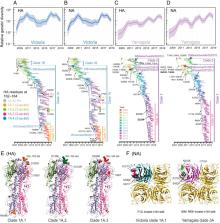
Influenza B viruses have circulated in humans for over 80 y, causing a significant disease burden. Two antigenically distinct lineages (“B/Victoria/2/87-like” and “B/Yamagata/16/88-like,” termed Victoria and Yamagata) emerged in the 1970s and have cocirculated since 2001. Since 2015 both lineages have shown unusually high levels of epidemic activity, the reasons for which are unclear. By analyzing over 12,000 influenza B virus genomes, we describe the processes enabling the long-term success and recent resurgence of epidemics due to influenza B virus. We show that following prolonged diversification, both lineages underwent selective sweeps across the genome and have subsequently taken alternate evolutionary trajectories to exhibit epidemic dominance, with no reassortment between lineages. Hemagglutinin deletion variants emerged concomitantly in multiple Victoria virus clades and persisted through epistatic mutations and interclade reassortment—a phenomenon previously only observed in the 1970s when Victoria and Yamagata lineages emerged. For Yamagata viruses, antigenic drift of neuraminidase was a major driver of epidemic activity, indicating that neuraminidase-based vaccines and cross-reactivity assays should be employed to monitor and develop robust protection against influenza B morbidity and mortality. Overall, we show that long-term diversification and infrequent selective sweeps, coupled with the reemergence of hemagglutinin deletion variants and antigenic drift of neuraminidase, are factors that contributed to successful circulation of diverse influenza B clades. Further divergence of hemagglutinin variants with poor cross-reactivity could potentially lead to circulation of 3 or more distinct influenza B viruses, further complicating influenza vaccine formulation and highlighting the urgent need for universal influenza vaccines.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology.
- Record: found
- Abstract: found
- Article: not found
Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics.
- Record: found
- Abstract: found
- Article: not found