- Record: found
- Abstract: found
- Article: not found
Mitigated NSAID-induced apoptotic and autophagic cell death with Smad7 overexpression
Read this article at

Abstract
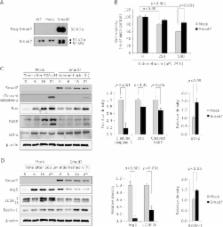
Non-steroidal anti-inflammatory drugs damaged gastrointestinal mucosa in cyclooxygenase-dependent and -independent pathway, among which apopototic or autophagic cell death in gastrointestinal cells might be one of key cytotoxic mechanisms responsible for NSAID-induced damages. Therefore, alleviating this cell death after NSAIDs can be a rescuing strategy. In this study, we explored the role of Smad7 on NSAID-induced cytotoxicity in gastric epithelial cells. Using RGM1 cells, we have compared biological changes between mock-transfected and Smad7-overexpressed cells. As results, significantly decreased cytotoxicity accompanied with decreased levels of cleaved caspase-3 and poly (ADP-ribose) polymerase, Bax, and autophagic vesicles concurrent with decreased expressions of autophagy protein 5 and microtubule-associated protein light chain 3B-II were noted in Smad7-overexpressed cells with indomethacin administration compared to mock-transfected cells. Contrast to mitigated apoptotic execution, anti-apoptotic Bcl-2 and Beclin-1 were significantly increased in Smad7-overexpressed cells compared to mock-transfected cells. Smad7 siRNA significantly reversed these protective actions of Smad7 against indomethacin, in which p38 mitogen-activated protein kinase was significantly intervened. Furthermore, indomethacin-induced Smad7 degradation through ubiquitin-proteasome pathway was relevant to increased cytotoxicity, while chloroquine as autophagy inhibitor significantly attenuated indomethacin-induced cytotoxicity through Smad7 preservation via repressed ubiquitination. Conclusively, either genetic overexpression or pharmacological induction of Smad7 significantly attenuated indomethacin-induced gastric cell damages.
Related collections
Most cited references25
- Record: found
- Abstract: not found
- Article: not found
Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs.
- Record: found
- Abstract: found
- Article: not found
Autophagy is activated for cell survival after endoplasmic reticulum stress.
- Record: found
- Abstract: found
- Article: not found