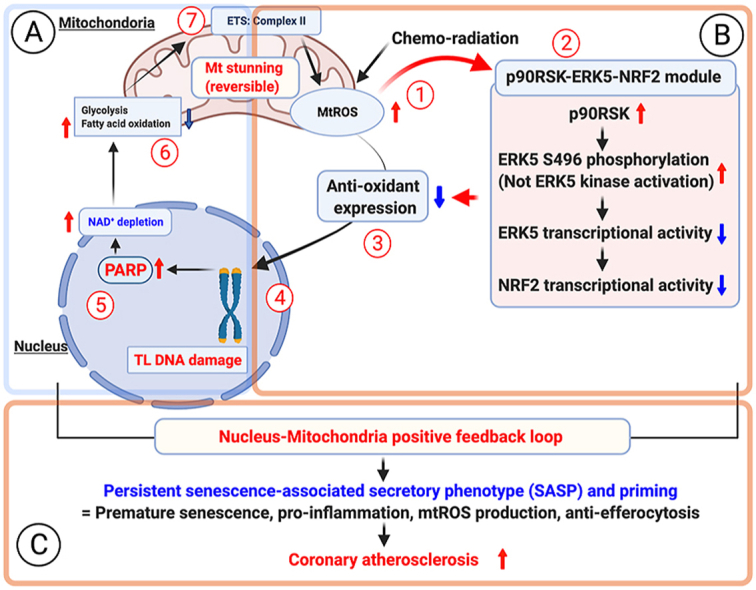
Introduction Cancer is a multistep disease in which cells acquire increasingly malignant phenotypes. These phenotypes are acquired in part by somatic mutations, which derange normal controls over cell proliferation (growth), survival, invasion, and other processes important for malignant tumorigenesis [1]. In addition, there is increasing evidence that the tissue microenvironment is an important determinant of whether and how malignancies develop [2,3]. Normal tissue environments tend to suppress malignant phenotypes, whereas abnormal tissue environments such at those caused by inflammation can promote cancer progression. Cancer development is restrained by a variety of tumor suppressor genes. Some of these genes permanently arrest the growth of cells at risk for neoplastic transformation, a process termed cellular senescence [4–6]. Two tumor suppressor pathways, controlled by the p53 and p16INK4a/pRB proteins, regulate senescence responses. Both pathways integrate multiple aspects of cellular physiology and direct cell fate towards survival, death, proliferation, or growth arrest, depending on the context [7,8]. Several lines of evidence indicate that cellular senescence is a potent tumor-suppressive mechanism [4,9,10]. Many potentially oncogenic stimuli (e.g., dysfunctional telomeres, DNA damage, and certain oncogenes) induce senescence [6,11]. Moreover, mutations that dampen the p53 or p16INK4a/pRB pathways confer resistance to senescence and greatly increase cancer risk [12,13]. Most cancers harbor mutations in one or both of these pathways [14,15]. Lastly, in mice and humans, a senescence response to strong mitogenic signals, such as those delivered by certain oncogenes, prevents premalignant lesions from progressing to malignant cancers [16–19]. Interestingly, some tumor cells retain the ability to senesce in response to DNA-damaging chemotherapy or p53 reactivation; in mice, this response arrests tumor progression [20–22]. Despite support for the idea that senescence is a beneficial anticancer mechanism, indirect evidence suggests that senescent cells can also be deleterious and might contribute to age-related pathologies [10,23–25]. The apparent paradox of contributing to both tumor suppression and aging is consistent with an evolutionary theory of aging, termed antagonistic pleiotropy [26]. Organisms generally evolve in environments that are replete with extrinsic hazards, and so old individuals tend to be rare in natural populations. Therefore, there is little selective pressure for tumor suppressor mechanisms to be effective well into old age; rather, these mechanisms need to be sufficiently effective only to ensure successful reproduction. Further, tumor suppressor mechanisms could in principle even be deleterious at advanced ages, as predicted by evolutionary antagonistic pleiotropy. Consistent with this view, senescent cells increase with age in mammalian tissues [27], and have been found at sites of age-related pathologies such as osteoarthritis and atherosclerosis [28–30]. Moreover, in mice, chronically active p53 both promotes cellular senescence and accelerates aging phenotypes [31,32]. How might senescent cells be deleterious? Senescent cells acquire many changes in gene expression, mostly documented as altered mRNA abundance, including increased expression of secreted proteins [33–41]. Some of these secreted proteins act in an autocrine manner to reinforce the senescence growth arrest [37,38,40,41]. Moreover, cell culture and mouse xenograft studies suggest that proteins secreted by senescent cells can promote degenerative or hyperproliferative changes in neighboring cells [35,39,42,43]. Thus, although the cell-autonomous senescence growth arrest suppresses cancer, factors secreted by senescent cells might have deleterious cell-nonautonomous effects that alter the tissue microenvironment. To date, a comprehensive analysis of the secretory profile of senescent cells is lacking, as is knowledge regarding how this profile varies with cell type or senescence inducer, or how it relates to the tumor suppressor proteins that control senescence. To fill these gaps in our knowledge, we modified antibody arrays to be quantitative and sensitive over a wide dynamic range and defined the senescence-associated secretory phenotype (SASP). We show that this phenotype is complex, containing elements associated with inflammation and tumorigenesis, and is induced only by genotoxic stress of sufficient magnitude to cause senescence. SASPs are expressed by senescent human fibroblasts and epithelial cells in culture. Moreover, epithelial tumor cells exposed to DNA-damaging chemotherapy senesce and express a SASP in vivo. The arrays allowed us to identify two new malignant phenotypes promoted by senescent cells (the epithelial–mesenchyme transition and invasiveness), and the SASP factors responsible for them (interleukin [IL]-6 and IL-8). Strikingly, the SASP was markedly amplified by oncogenic RAS or loss of p53 function. Our results identify a mechanism by which p53 acts as a cell-nonautonomous tumor suppressor, and RAS as a cell-nonautonomous oncogene, and provide a novel framework for understanding how age-related cancers might progress. Results Senescence-Associated Secretory Phenotypes Expressed by Human Fibroblasts To determine whether tissue of origin, donor age, or genotype affected secretory phenotypes, we first studied five human fibroblast strains, derived from embryonic lung (WI-38, IMR-90), neonatal foreskin (BJ, HCA2), or adult breast (hBF184). We cultured the cells under standard conditions, and either atmospheric (∼20%) O2 or 3% O2, which is more physiological [44]. We made presenescent (PRE) cultures (>80% of cells capable of proliferation) quiescent by growing the cells to confluence in order to compare them to nondividing senescent (SEN) cultures ( 0.75) in human senescent cells cultured in 3% versus 20% O2. By contrast, the ambient O2 level strongly affects the secretory phenotype of mouse senescent cells (J. P. Coppe, C. K. Patil, F. Rodier, A. Krtolica, S. Parrinello, et al., unpublished data). We verified the secretion levels of several SASP proteins by ELISAs (Figure S1 and Text S1). Further, because secretion increased greater than 10-fold for some SASP factors, we could verify up-regulation by intracellular immunostaining. For example, IL-6 and IL-8 were barely visible in PRE cells but clearly detectable in SEN cells (Figure 1B, Figure S2, and Text S1). We performed the immunostaining on cells in 10% serum, which allowed us to rule out the possibility that the SASP was a senescence-specific response to the serum-free incubation needed to collect CM. Moreover, the SASPs of SEN cells induced by REP and XRA were highly correlated (r > 0.9; Figure 1C), indicating that the phenotype was not specific to one senescence inducer. The secretory profiles of fibroblast strains from the same tissues (e.g., BJ and HCA2 from neonatal foreskin; and IMR90 and Wi-38 from fetal lung) were highly correlated (Datasets S3 and S4). In subsequent figures, data from these related cell strains, as well as from REP and XRA samples from cells of the same type, were pooled and averaged in order to simplify the display. Because REP and XRA induce senescence primarily by causing genomic damage (from telomere shortening and DNA breaks, respectively), we asked whether the SASP was a primary DNA damage response. We irradiated cells using either 0.5 or 10 Gy. As expected, both doses initiated a DNA damage response, as determined by p53 stabilization and phosphorylation (see Figure S3 and Text S1). However, cells that received 0.5 Gy transiently arrested growth for only 24–48 h before resuming growth, whereas cells that received 10 Gy underwent a permanent senescence growth arrest (Figure 1D). Antibody arrays performed on CM collected between 2 and 10 d after irradiation showed that only 10 Gy induced a SASP (Figure 1D and Figure S3). Moreover, cells that senesced owing to DNA damage developed the SASP slowly, requiring 4–7 d after irradiation before expressing a robust SASP. These findings indicate the SASP is not a DNA damage response per se. However, it is induced by DNA damage of sufficient magnitude to cause senescence, after which it requires several days to develop. We also determined that proteins comprising the SASP were, in general, up-regulated at the level of mRNA abundance (Figure 1E and 1F, red symbols and line; Figure S4, and Text S1). However, for detectable proteins that showed little or no senescence-associated change in secretion, mRNA levels were a poor predictor of secreted protein levels (Figure 1F, blue symbols and line; and Figure S4). Thus, antibody arrays provide a more accurate assessment of the senescence-associated secretory signature than mRNA profiling. SASPs of Human Epithelial Cells To determine whether the SASP is limited to fibroblasts, we studied the secretory activity of epithelial cells. Normal human prostate epithelial cells (PrECs) underwent a classic senescence growth arrest in response to X-irradiation (see Table S1). We collected CM from PRE and SEN PrECs, and analyzed the factors secreted by these cells using antibody arrays. Normal PrECs expressed a robust SASP upon senescence (Figure 2A and Datasets S5–S8). Like fibroblasts, SEN PrECs secreted many factors at significantly higher levels compared to PRE PrECs. To compare the SASPs of normal human epithelial and stromal cells senesced under similar conditions, we analyzed factors that showed a significant change (p 66% overlap between normal fibroblasts and normal epithelial cells. More specifically, both SASPs included inflammatory or immune factors such as IL-6, IL-8, or MCP-1, growth modulators such as GRO and IGFBP-2, cell survival regulators such as OPG or sTNF RI, and shed surface molecules such as uPAR or ICAM-1 (Figure 2A). Not surprising, there were also differences between the SASPs of fibroblasts and PrECs. In contrast to fibroblasts, three factors (Acrp30, BTC, and IGFBP-6) were significantly down-regulated by SEN in PrECs. Moreover, IL-1α or HGF were SASP factors unique to either normal epithelial SASP or normal fibroblast SASP, respectively. This result indicates that the SASP is not limited to normal stromal cells, and that a substantial overlap between normal senescent cells of different tissue origins exists. Figure 2 SASP of Human Epithelial Cells (A) Soluble factors secreted by the indicated normal cell type (epithelial vs. stromal) were detected by antibody arrays and analyzed as described in the text, Materials and Methods, and Datasets S5–S8. Normal prostate epithelial cells (PrECs) were induced to senesce by 10 Gy irradiation, and CM from the PRE and SEN cells were analyzed. The SASP of PrECs was compared side by side to the SASP of SEN(XRA) fibroblasts (WI-38, IMR-90, HCA-2, and BJ). The PRE values for each cell type served as the baseline. Signals higher than baseline are displayed in yellow; signals below baseline are displayed in blue. The heat map key (right) indicates log2-fold changes from the baseline. p-Values were calculated by the Student t-test, and are given to the right of the heat map. ns = not significant (p > 0.05) and defines non-SASP factors. (B) The log2-fold changes for all 120 proteins detected by the antibody arrays were plotted for SEN(XRA) PrECs and SEN(XRA) fibroblasts, relative to their PRE baseline. Seventy-nine secreted factors (66%) followed the same regulatory trend (in red). The remaining factors were not coregulated (depicted in blue). (C) Soluble factors produced by the indicated normal or transformed prostate epithelial cells were analyzed by antibody arrays and the results displayed as described for Figure 1A. For each cell strain or cell line, PRE and SEN signals were averaged and used as the baseline. Signals higher than the baseline are shown in yellow; signals below baseline are displayed in blue. An asterisk (*) indicates SASP factors that are conserved between all fibroblasts (Figure 1A) and all epithelial cells. Some tumor cells retain the ability to senesce in response to DNA damage, including DNA-damaging chemotherapy [20–22]. We therefore asked whether prostate cancer cells also developed a SASP. We studied three prostate tumor cell lines (BPH1 [45], RWPE1 [46], and PC3 [47], which differ in their degree of malignancy as follows: PC3 > RWPE1 > BPH1). As with normal epithelial cells (PrECs), we induced senescence by XRA, and analyzed CM using antibody arrays (Datasets S5–S8). SEN epithelial cells secreted significantly higher levels of numerous proteins compared to PRE counterparts (Figure 2C). The SASPs of prostate epithelial cells showed striking overlap between normal and transformed cells (Figure 2C and Dataset S8), and there was also striking similarities between the SASP of fibroblasts and all epithelial cells, transformed and not transformed (Figure 2C, asterisks indicate common secreted proteins, and Datasets S5–S8). Twenty-four proteins were shared between the SASPs of all fibroblasts (Figure 1A) and all epithelial cells (Figure 2C); this overlap was highly significant relative to the overlap predicted from chance (p = ∼10−5; see Materials and Methods). We conclude that normal fibroblasts and both normal and transformed epithelial cells can develop SASPs that significantly overlap, displaying many common, but also some distinct, features. Chemotherapy-Induced SASPs Occur In Vivo Many human tumor cells retain the ability to senesce, in culture and in vivo, in response to DNA-damaging chemotherapeutic agents [48,49]. Epithelial cell lines, as well as normal fibroblasts, underwent senescence in culture in response to mitoxantrone (MIT) (see Table S1), a topoisomerase 2β inhibitor that causes DNA breaks and is used to treat prostate cancer [50]. Antibody arrays (Figure 3A and Datasets S5–S8) and ELISAs for IL-6, IL-8, and GRO-α (Figure S1) showed that MIT induced a SASP that correlated well (r = 0.89) with the XRA-induced SASP (Figure 3A). Figure 3 Chemotherapy-Induced SASP in Culture and In Vivo (A) Overall correlation between XRA and mitoxantrone (MIT)-induced SASPs for all three prostate epithelial cancer cell lines (BPH-1, RWPE-1, and PC-3). Correlations for the individual cell lines are given in the table to the right. The senescence inducer (XRA or MIT) is given in parentheses. (B–E) Human tumor cells were isolated from biopsies obtained from the same patient before MIT chemotherapy and from prostate tissue following prostatectomy after MIT chemotherapy. Laser captured cells were analyzed by quantitative RT-PCR for the mRNAs encoding the indicated proteins, as described in Materials and Methods and Text S1. The results are displayed on a log10 scale indicating the values before (horizontal or x-axis) compared to after (vertical or y-axis) chemotherapy (top left panel in [B]). Each black dot in (B, C, and D) represents the results obtained from a single patient. The average values for all patients before versus after chemotherapy are indicated by a red dot (B–D); these values are also represented as a heat map in (E). (B) Values for mRNAs encoding proteins associated with senescence (p16 and p21) and proliferation (cyclin A, MCM-3, and PCNA). (C) Values for mRNAs encoding SASP-associated proteins (IL-6, IL-8, GM-CSF, GRO-α, IGFBP-2, and IL-1β). (D) Value for an mRNA encoding a non-SASP–associated protein (IL-2). (E) Averages for the values shown in (B–D). Overall p-values, determined by the Student t-test, and number of paired samples (or patients) analyzed for each mRNA are given to the right of the heat map. Signals higher than the prechemotherapy baseline are shown in red; signals below baseline are displayed in green. The finding that human prostatic tumor cells express a SASP in response to MIT in culture allowed us to determine whether MIT induced a SASP in vivo. We laser captured approximately 1,000 tumor epithelial cells in biopsies from human prostate cancer patients before MIT chemotherapy and in tissues removed after chemotherapy and prostatectomy [50]. By microscopic inspection, the captured cells were devoid of stromal cells and leukocytes. Since mRNA and secreted protein levels correlated well for significantly up-regulated SASP factors (Figure 1E and 1F and Figure S4), we used quantitative PCR to quantify mRNAs encoding senescence and proliferation markers and SASP factors. After chemotherapy, most of the tumors contained significantly higher levels of p16INK4a and p21 mRNAs, which are typically up-regulated in senescent cells (Figure 3B). They also contained significantly lower levels of proliferation-associated mRNAs encoding cyclin A, MCM-3, and PCNA (Figure 3B). These results suggest that MIT induced tumor cells to senesce in vivo. Importantly, most of the tumors contained significantly higher levels of mRNAs encoding the SASP components IL-6, IL-8, GM-CSF, GRO-α, IGFBP-2, and IL-1β (Figure 3C). However, the levels of mRNA encoding IL-2, which is not a SASP component, did not significantly change on average (Figure 3D). These findings (summarized in Figure 3E) suggest that the SASP is not limited to cultured cells, but also occurs when human cells senesce in vivo. SASPs Induce Epithelial–Mesenchyme Transitions and Invasiveness The epithelial–mesenchymal transition (EMT) confers invasive and metastatic properties on epithelial cells, and is an important step in cancer progression that presages the conversion of carcinomas in situ to potentially fatal invasive cancers [51,52]. We found that the fibroblast SASP induced a classic EMT in two nonaggressive human breast cancer cell lines (T47D and ZR75.1). Secreted factors from SEN, but not PRE, fibroblasts caused dose-dependent epithelial cell scattering, a mesenchymal characteristic (Figure 4A). Moreover, immunostaining showed that PRE CM preserved surface-associated β-catenin and E-cadherin, and strong cytokeratin 8/18 expression (Figure 4B), and western analysis showed that PRE CM preserved low expression of vimentin (see below). These features are epithelial characteristics frequently retained by nonaggressive cells [51,52]. By contrast, CM from SEN cells markedly decreased overall and cell surface β-catenin and E-cadherin and reduced cytokeratin expression (Figure 4B), consistent with a mesenchymal transition. Further, SEN CM down-regulated the tight junction protein claudin-1, leaving the remaining protein localized primarily to the nucleus (Figure 4B), a hallmark of an EMT and a feature of metastatic but not primary tumors [53]. Finally, SEN CM increased vimentin expression (see below), another mesenchymal marker and hallmark of an EMT [52]. Figure 4 Novel SASP Biological Activities and Key Factors (A) T47D and ZR75.1 cells were incubated for 2 d with CM from PRE fibroblasts, or SEN fibroblasts induced by XRA. The cells were photographed under phase contrast, or analyzed for cluster size using an automated Cellomix imager and software. Smaller cluster or clump sizes (pixel2) indicate greater scattering. The senescence inducer is given in parentheses. Quadruple asterisks (****) indicate p GSE) versus SEN(REP) in WI-38) and Datasets S13–S16). This finding indicates that p53 is not required to maintain an established SASP. Figure 6 p53 Restrains the SASP (A) CM containing factors secreted by the indicted cells were analyzed by antibody arrays and displayed, using PRE CM as the baseline. We pooled data from cells of the same genotype (p53 wild type or p53 deficient) under the same culture conditions. SEN indicates pooled data from cells originating from the same tissue (WI-38, IMR-90 from embryonic lung; and HCA-2, BJ from neonatal foreskin) and induced to senesce by REP or XRA. Pooling and averaging of highly correlated samples was performed as described for Figure 5, and details of the data processing are provided in Datasets S13–S16. The top four rows are the same top four rows in Figure 5A and are included to serve as a visual reference. The senescence inducer is given in parentheses. p53 status is indicated as either wild type (wt) or deficient (d) owing to either GSE22 expression or expression of an shRNA against p53. Manipulations are indicated in sequence, separated by a greater than symbol (>). The heat map key (right) indicates the log2-fold changes. Signals higher than the baseline are shown in yellow; signals below baseline are displayed in blue. Comparison between rows is accurately illustrated in (B) and (C) in which each genetically manipulated cell type is compared to its appropriate control baseline. (B) Log2-fold values for SASP factors that are significantly increased, or significantly and uniquely (as indicated by double asterisks [**]) elevated, in CM from SEN cells made p53 deficient by GSE22, using untreated wild-type SEN values as the baseline. Green indicates WI-38 cells made senescent by XRA, after which p53 was subsequently inactivated by expressing GSE22 using a lentivirus; these cells do not resume proliferation (“unreverted”) upon p53 inactivation (see Figure S6). Blue indicates WI-38 in which p16 was inactivated by an shRNA, induced to senesce by XRA, then infected with the GSE22-expressing lentivirus; these cells do revert (REV) after p53 inactivation. Pink indicates HCA2 cells made SEN by XRA, then infected with GSE22 lentivirus; these cells also revert after p53 inactivation. (C) Log2-fold values for SASP factors that are significantly increased, or significantly and uniquely (as indicated by double asterisks [**]) elevated, in CM from cells made p53-deficient (by GSE22 expression), then induced to senesce by REP, XRA, or RAS. Red indicates WI-38 and IMR90 (averaged) cells expressing GSE22, then induced to senesce by XRA or REP, using cells made SEN by XRA or REP as a baseline. Gray indicates WI-38, IMR-90, and HCA2 (averaged) expressing GSE22, then made senescent by RAS, using SEN by RAS as a baseline. (D) WI-38 cells expressing GSE22 were induced to senesce by XRA and then immunostained for the SASP proteins IL-6 and IL-8, the senescence marker p16INK4a, and p53, which accumulates in the presence of GSE22. (E) Comparative graphical representation of the secretory profiles of cells made senescent by XRA or REP (dotted line), RAS (black line), p53 inactivation (GSE22) followed by XRA or REP (blue line), or p53 inactivation (GSE22) followed by RAS (red line). The increased slopes (as indicated by the arrow)indicate amplified SASPs. (F) Hierarchical cluster analysis of all the cells analyzed in (A), plus the SASP induced by RAS (see Figure 5). RAS status is indicated as either wild type (wt) or oncogenic (o) owing to expression of Ha-RASv12. (G) WI-38 cells with wild-type (wt) or inactive (GSE) p53 were irradiated or induced to express oncogenic RAS (RAS), and CM was collected 4 or 10 d later. Soluble factors were analyzed by antibody arrays and displayed as described in Figure 1D, using PRE CM as the baseline (black column on the left; see also Figure S5C for details). Signals higher than baseline are shown in yellow; signals below baseline are in blue. n/a, not applicable. (H) Log2-fold values for prostate epithelial cell SASP factors that are significantly or uniquely (as indicated by double asterisks [**]) elevated in CM from p53-deficient cancer cells (PC3, BPH1, and RWPE1) that were induced to senesce by XRA, compared to primary p53 wild-type cells (PrECs) that were induced to senesce by XRA. Figure 7 Biological Activities of the Amplified SASP (A) T47D and ZR75.1 cells were incubated with the indicated CM for 3 d and then analyzed for cell scattering, immunostained for the indicated proteins, and analyzed for vimentin and actin levels by western blotting. Controls for the immunofluorescence from the same individual experiment are shown in Figure 4B. The senescence inducer is given in parentheses. p53 status was either wild type or deficient (GSE). Manipulations are indicated in sequence, separated by >. (B) Epithelial cells were incubated with CM from the indicated WI-38 cells and assayed for invasion as described in Materials and Methods and Figure 3C. Double asterisks (**) indicate p 0.95). At 4 d post 10 Gy irradiation, cells harboring a wild-type p53 pathway are still very similar to their PRE counterpart (correlation > 0.95), whereas at 10 d post 10 Gy irradiation, cells are very dissimilar to PRE (correlation < 0 ; they have developed a SASP). The clustering analysis also shows that cells that senesced in the absence of p53 function or due to oncogenic RAS overexpression resemble more each other than cells that senesced with a wild-type p53 background, suggesting that the loss of p53 and the gain of oncogenic RAS have similar dominant effects over SASP development and establishment. (142 KB PDF) Click here for additional data file. Figure S6 Growth Reversion of SEN Cells after p53 Inactivation SEN(REP) and SEN(XRA) WI-38 cells were monitored for cell growth for 20 d before infection with lenti-GSE (rectangle). Cell number was subsequently monitored for an additional 30 d thereafter. Because SEN WI-38 cells express p16, p53 inactivation by GSE does not revert the SEN growth arrest. Cells that do not express p16 at SEN (shp16-expressing WI-38 or unmodified HCA2 cells) were similarly monitored and infected. In contrast to SEN WI-38 cells, p16-deficient cells resumed growth (reverted) after p53-inactivation and proliferated for at least the ensuing 30 d. (79 KB PDF) Click here for additional data file. Dataset S1 Computational Analysis of Antibody Array Data Presented in Figure 1 (Human Fibroblasts; SEN(XRA) and SEN(REP)): Part 1 (31 KB DOC) Click here for additional data file. Dataset S2 Computational Analysis of Antibody Array Data Presented in Figure 1 (Human Fibroblasts; SEN(XRA) and SEN(REP)): Part 2 (28 KB XLS) Click here for additional data file. Dataset S3 Computational Analysis of Antibody Array Data Presented in Figure 1 (Human Fibroblasts; SEN(XRA) and SEN(REP)): Part 3 (142 KB XLS) Click here for additional data file. Dataset S4 Computational Analysis of Antibody Array Data Presented in Figure 1 (Human Fibroblasts; SEN(XRA) and SEN(REP)): Part 4 (162 KB XLS) Click here for additional data file. Dataset S5 Computational Analysis of Antibody Array Data Presented in Figure 2 and Figure 3 (Human Epithelial Cells; SEN(XRA)): Part 1 (31 KB DOC) Click here for additional data file. Dataset S6 Computational Analysis of Antibody Array Data Presented in Figure 2 and Figure 3 (Human Epithelial Cells; SEN(XRA)): Part 2 (39 KB XLS) Click here for additional data file. Dataset S7 Computational Analysis of Antibody Array Data Presented in Figure 2 and Figure 3 (Human Epithelial Cells; SEN(XRA)): Part 3 (78 KB XLS) Click here for additional data file. Dataset S8 Computational Analysis of Antibody Array Data Presented in Figure 2 and Figure 3 (Human Epithelial Cells; SEN(XRA)): Part 4 (159 KB XLS) Click here for additional data file. Dataset S9 Computational Analysis of Antibody Array Data Presented in Figure 5 (Oncogene-Induced Senescence): Part 1 (31 KB DOC) Click here for additional data file. Dataset S10 Computational Analysis of Antibody Array Data Presented in Figure 5 (Oncogene-Induced Senescence): Part 2 (19 KB XLS) Click here for additional data file. Dataset S11 Computational Analysis of Antibody Array Data Presented in Figure 5 (Oncogene-Induced Senescence): Part 3 (90 KB XLS) Click here for additional data file. Dataset S12 Computational Analysis of Antibody Array Data Presented in Figure 5 (Oncogene-Induced Senescence): Part 4 (61 KB XLS) Click here for additional data file. Dataset S13 Computational Analysis of Antibody Array Data Presented in Figure 6 (p53-Deficient Senescence): Part 1 (31 KB DOC) Click here for additional data file. Dataset S14 Computational Analysis of Antibody Array Data Presented in Figure 6 (p53-Deficient Senescence): Part 2 (25 KB XLS) Click here for additional data file. Dataset S15 Computational Analysis of Antibody Array Data Presented in Figure 6 (p53-Deficient Senescence): Part 3 (133 KB XLS) Click here for additional data file. Dataset S16 Computational Analysis of Antibody Array Data Presented in Figure 6 (p53-Deficient Senescence): Part 4 (81 KB XLS) Click here for additional data file. Table S1 Presenescent and Senescent Cells Characteristics Labeling index and senescence-associated beta-galactosidase (SA-bGal) staining of human fibroblasts and human prostate epithelial cells in vitro. (194 KB DOC) Click here for additional data file. Table S2 Complete Catalog of Entrez Gene IDs for All Proteins Corresponding to Antibodies on the Arrays (61 KB XLS) Click here for additional data file. Text S1 Supplemental Methods (42 KB DOC) Click here for additional data file. Text S2 Computational Processing, Analysis, and Validation of Antibody Arrays Digitization and quantification of antibody arrays; numerical and statistical methods; linearity; accuracy; and reliability. (289 KB DOC) Click here for additional data file.