- Record: found
- Abstract: found
- Article: found
Membrane Transport Proteins in Osteoclasts: The Ins and Outs
Read this article at
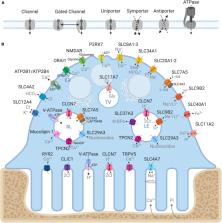
Abstract
During bone resorption, the osteoclast must sustain an extraordinarily low pH environment, withstand immense ionic pressures, and coordinate nutrient and waste exchange across its membrane to sustain its unique structural and functional polarity. To achieve this, osteoclasts are equipped with an elaborate set of membrane transport proteins (pumps, transporters and channels) that serve as molecular ‘gatekeepers’ to regulate the bilateral exchange of ions, amino acids, metabolites and macromolecules across the ruffled border and basolateral domains. Whereas the importance of the vacuolar-ATPase proton pump and chloride voltage-gated channel 7 in osteoclasts has long been established, comparatively little is known about the contributions of other membrane transport proteins, including those categorized as secondary active transporters. In this Special Issue review, we provide a contemporary update on the ‘ins and outs’ of membrane transport proteins implicated in osteoclast differentiation, function and bone homeostasis and discuss their therapeutic potential for the treatment of metabolic bone diseases.
Related collections
Most cited references229
- Record: found
- Abstract: found
- Article: not found
A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function.
- Record: found
- Abstract: found
- Article: not found