- Record: found
- Abstract: found
- Article: not found
PIP 2-Binding Site in Kir Channels: Definition by Multiscale Biomolecular Simulations†
Read this article at

Abstract

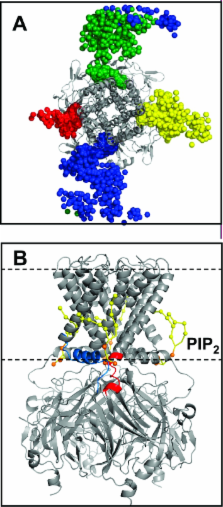
Phosphatidylinositol bisphosphate (PIP 2) is an activator of mammalian inwardly rectifying potassium (Kir) channels. Multiscale simulations, via a sequential combination of coarse-grained and atomistic molecular dynamics, enabled exploration of the interactions of PIP 2 molecules within the inner leaflet of a lipid bilayer membrane with possible binding sites on Kir channels. Three Kir channel structures were investigated: X-ray structures of KirBac1.1 and of a Kir3.1−KirBac1.3 chimera and a homology model of Kir6.2. Coarse-grained simulations of the Kir channels in PIP 2-containing lipid bilayers identified the PIP 2-binding site on each channel. These models of the PIP 2−channel complexes were refined by conversion to an atomistic representation followed by molecular dynamics simulation in a lipid bilayer. All three channels were revealed to contain a conserved binding site at the N-terminal end of the slide (M0) helix, at the interface between adjacent subunits of the channel. This binding site agrees with mutagenesis data and is in the proximity of the site occupied by a detergent molecule in the Kir chimera channel crystal. Polar contacts in the coarse-grained simulations corresponded to long-lived electrostatic and H-bonding interactions between the channel and PIP 2 in the atomistic simulations, enabling identification of key side chains.
Related collections
Most cited references44
- Record: found
- Abstract: found
- Article: not found
A multiscale coarse-graining method for biomolecular systems.
- Record: found
- Abstract: found
- Article: not found
Crystal structure of the potassium channel KirBac1.1 in the closed state.
- Record: found
- Abstract: found
- Article: not found