- Record: found
- Abstract: found
- Article: found
Gut dysbiosis and impairment of immune system homeostasis in perinatally-exposed mice to Bisphenol A precede obese phenotype development
Read this article at
Abstract
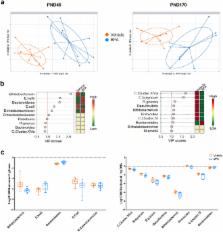
Epidemiology evidenced the Bisphenol A (BPA), a chemical found in daily consumer products, as an environmental contributor to obesity and type II diabetes (T2D) in Humans. However, the BPA-mediated effects supporting these metabolic disorders are still unknown. Knowing that obesity and T2D are associated with low-grade inflammation and gut dysbiosis, we performed a longitudinal study in mice to determine the sequential adverse effects of BPA on immune system and intestinal microbiota that could contribute to the development of metabolic disorders. We observed that perinatal exposure to BPA (50 µg/kg body weight/day) induced intestinal and systemic immune imbalances at PND45, through a decrease of Th1/Th17 cell frequencies in the lamina propria concomitant to an increase of splenic Th1/Th17 immune responses. These early effects are associated with an altered glucose sensitivity, a defect of IgA secretion into faeces and a fall of faecal bifidobacteria relative to control mice. Such BPA-mediated events precede infiltration of pro-inflammatory M1 macrophages in gonadal white adipose tissue appearing with ageing, together with a decreased insulin sensitivity and an increased weight gain. Our findings provide a better understanding of the sequential events provoked by perinatal exposure to BPA that could support metabolic disorder development in later life.
Related collections
Most cited references34
- Record: found
- Abstract: found
- Article: not found
Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans.
- Record: found
- Abstract: found
- Article: not found
Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study.
- Record: found
- Abstract: found
- Article: not found