- Record: found
- Abstract: found
- Article: found
Propofol inhibits prokaryotic voltage-gated Na + channels by promoting activation-coupled inactivation
Read this article at
Abstract
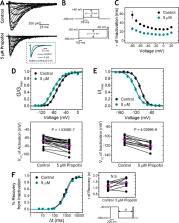
Despite extensive use in clinical practice, the mechanisms of propofol action on sodium channels are not fully understood. Yang et al. incorporate complementary biophysical approaches (electrophysiology and molecular dynamics simulations) to demonstrate that propofol inhibits two prokaryotic voltage-gated sodium channels, NaChBac and NavMs, by modulating both activation and inactivation gating.
Abstract
Propofol is widely used in the clinic for the induction and maintenance of general anesthesia. As with most general anesthetics, however, our understanding of its mechanism of action remains incomplete. Local and general anesthetics largely inhibit voltage-gated Na + channels (Navs) by inducing an apparent stabilization of the inactivated state, associated in some instances with pore block. To determine the biophysical and molecular basis of propofol action in Navs, we investigated NaChBac and NavMs, two prokaryotic Navs with distinct voltage dependencies and gating kinetics, by whole-cell patch clamp electrophysiology in the absence and presence of propofol at clinically relevant concentrations (2–10 µM). In both Navs, propofol induced a hyperpolarizing shift of the pre-pulse inactivation curve without any significant effects on recovery from inactivation at strongly hyperpolarized voltages, demonstrating that propofol does not stabilize the inactivated state. Moreover, there was no evidence of fast or slow pore block by propofol in a non-inactivating NaChBac mutant (T220A). Propofol also induced hyperpolarizing shifts of the conductance-voltage relationships with negligible effects on the time constants of deactivation at hyperpolarized voltages, indicating that propofol does not stabilize the open state. Instead, propofol decreases the time constants of macroscopic activation and inactivation. Adopting a kinetic scheme of Nav gating that assumes preferential closed-state recovery from inactivation, a 1.7-fold acceleration of the rate constant of activation and a 1.4-fold acceleration of the rate constant of inactivation were sufficient to reproduce experimental observations with computer simulations. In addition, molecular dynamics simulations and molecular docking suggest that propofol binding involves interactions with gating machinery in the S4–S5 linker and external pore regions. Our findings show that propofol is primarily a positive gating modulator of prokaryotic Navs, which ultimately inhibits the channels by promoting activation-coupled inactivation.
Related collections
Most cited references81
- Record: found
- Abstract: found
- Article: not found
General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal.
- Record: found
- Abstract: found
- Article: not found
Voltage sensor of Kv1.2: structural basis of electromechanical coupling.
- Record: found
- Abstract: found
- Article: not found