- Record: found
- Abstract: found
- Article: found
Whole-Genome Sequencing for Routine Pathogen Surveillance in Public Health: a Population Snapshot of Invasive Staphylococcus aureus in Europe
Read this article at
ABSTRACT
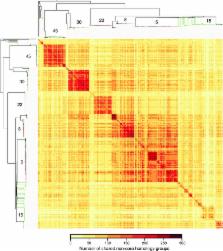
The implementation of routine whole-genome sequencing (WGS) promises to transform our ability to monitor the emergence and spread of bacterial pathogens. Here we combined WGS data from 308 invasive Staphylococcus aureus isolates corresponding to a pan-European population snapshot, with epidemiological and resistance data. Geospatial visualization of the data is made possible by a generic software tool designed for public health purposes that is available at the project URL ( http://www.microreact.org/project/EkUvg9uY?tt=rc). Our analysis demonstrates that high-risk clones can be identified on the basis of population level properties such as clonal relatedness, abundance, and spatial structuring and by inferring virulence and resistance properties on the basis of gene content. We also show that in silico predictions of antibiotic resistance profiles are at least as reliable as phenotypic testing. We argue that this work provides a comprehensive road map illustrating the three vital components for future molecular epidemiological surveillance: (i) large-scale structured surveys, (ii) WGS, and (iii) community-oriented database infrastructure and analysis tools.
IMPORTANCE
The spread of antibiotic-resistant bacteria is a public health emergency of global concern, threatening medical intervention at every level of health care delivery. Several recent studies have demonstrated the promise of routine whole-genome sequencing (WGS) of bacterial pathogens for epidemiological surveillance, outbreak detection, and infection control. However, as this technology becomes more widely adopted, the key challenges of generating representative national and international data sets and the development of bioinformatic tools to manage and interpret the data become increasingly pertinent. This study provides a road map for the integration of WGS data into routine pathogen surveillance. We emphasize the importance of large-scale routine surveys to provide the population context for more targeted or localized investigation and the development of open-access bioinformatic tools to provide the means to combine and compare independently generated data with publicly available data sets.
Related collections
Most cited references41
- Record: found
- Abstract: found
- Article: not found
eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data.
- Record: found
- Abstract: found
- Article: not found
The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA).
- Record: found
- Abstract: found
- Article: not found