- Record: found
- Abstract: found
- Article: found
A random forest based computational model for predicting novel lncRNA-disease associations
Read this article at
Abstract
Background
Accumulated evidence shows that the abnormal regulation of long non-coding RNA (lncRNA) is associated with various human diseases. Accurately identifying disease-associated lncRNAs is helpful to study the mechanism of lncRNAs in diseases and explore new therapies of diseases. Many lncRNA-disease association (LDA) prediction models have been implemented by integrating multiple kinds of data resources. However, most of the existing models ignore the interference of noisy and redundancy information among these data resources.
Results
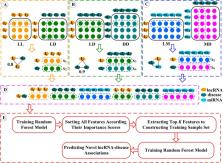
To improve the ability of LDA prediction models, we implemented a random forest and feature selection based LDA prediction model (RFLDA in short). First, the RFLDA integrates the experiment-supported miRNA-disease associations (MDAs) and LDAs, the disease semantic similarity (DSS), the lncRNA functional similarity (LFS) and the lncRNA-miRNA interactions (LMI) as input features. Then, the RFLDA chooses the most useful features to train prediction model by feature selection based on the random forest variable importance score that takes into account not only the effect of individual feature on prediction results but also the joint effects of multiple features on prediction results. Finally, a random forest regression model is trained to score potential lncRNA-disease associations. In terms of the area under the receiver operating characteristic curve (AUC) of 0.976 and the area under the precision-recall curve (AUPR) of 0.779 under 5-fold cross-validation, the performance of the RFLDA is better than several state-of-the-art LDA prediction models. Moreover, case studies on three cancers demonstrate that 43 of the 45 lncRNAs predicted by the RFLDA are validated by experimental data, and the other two predicted lncRNAs are supported by other LDA prediction models.
Related collections
Most cited references52
- Record: found
- Abstract: found
- Article: not found
A new method to measure the semantic similarity of GO terms.

- Record: found
- Abstract: found
- Article: found
Long non-coding RNAs and complex diseases: from experimental results to computational models

- Record: found
- Abstract: found
- Article: found