- Record: found
- Abstract: found
- Article: found
Depressed neuromuscular transmission causes weakness in mice lacking BK potassium channels
Read this article at
Abstract
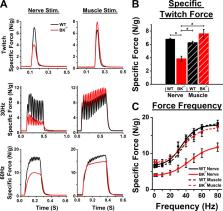
Mice lacking BK potassium channels have weakness with stimulation of peripheral nerve, but not muscle, which is caused by a defect in neuromuscular transmission. Prolonging the motor neuron action potential fully normalizes in vivo strength.
Abstract
Mice lacking functional large-conductance voltage- and Ca 2+-activated K + channels (BK channels) are viable but have motor deficits including ataxia and weakness. The cause of weakness is unknown. In this study, we discovered, in vivo, that skeletal muscle in mice lacking BK channels (BK −/−) was weak in response to nerve stimulation but not to direct muscle stimulation, suggesting a failure of neuromuscular transmission. Voltage-clamp studies of the BK −/− neuromuscular junction (NMJ) revealed a reduction in evoked endplate current amplitude and the frequency of spontaneous vesicle release compared with WT littermates. Responses to 50-Hz stimulation indicated a reduced probability of vesicle release in BK −/− mice, suggestive of lower presynaptic Ca 2+ entry. Pharmacological block of BK channels in WT NMJs did not affect NMJ function, surprisingly suggesting that the reduced vesicle release in BK −/− NMJs was not due to loss of BK channel–mediated K + current. Possible explanations for our data include an effect of BK channels on development of the NMJ, a role for BK channels in regulating presynaptic Ca 2+ current or the effectiveness of Ca 2+ in triggering release. Consistent with reduced Ca 2+ entry or effectiveness of Ca 2+ in triggering release, use of 3,4-diaminopyridine to widen action potentials normalized evoked release in BK −/− mice to WT levels. Intraperitoneal application of 3,4-diaminopyridine fully restored in vivo nerve-stimulated muscle force in BK −/− mice. Our work demonstrates that mice lacking BK channels have weakness due to a defect in vesicle release at the NMJ.
Related collections
Most cited references53
- Record: found
- Abstract: found
- Article: not found
Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.
- Record: found
- Abstract: found
- Article: not found
Short-term synaptic plasticity.
- Record: found
- Abstract: found
- Article: not found