- Record: found
- Abstract: found
- Article: found
Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy
Read this article at
Abstract
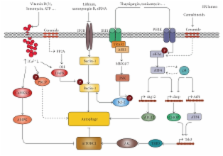
Different physiological and pathological conditions can perturb protein folding in the endoplasmic reticulum, leading to a condition known as ER stress. ER stress activates a complex intracellular signal transduction pathway, called unfolded protein response (UPR). The UPR is tailored essentially to reestablish ER homeostasis also through adaptive mechanisms involving the stimulation of autophagy. However, when persistent, ER stress can switch the cytoprotective functions of UPR and autophagy into cell death promoting mechanisms. Recently, a variety of anticancer therapies have been linked to the induction of ER stress in cancer cells, suggesting that strategies devised to stimulate its prodeath function or block its prosurvival function, could be envisaged to improve their tumoricidial action. A better understanding of the molecular mechanisms that determine the final outcome of UPR and autophagy activation by chemotherapeutic agents, will offer new opportunities to improve existing cancer therapies as well as unravel novel targets for cancer treatment.
Related collections
Most cited references148
- Record: found
- Abstract: found
- Article: not found
Autophagy is activated for cell survival after endoplasmic reticulum stress.
- Record: found
- Abstract: found
- Article: not found
The role of autophagy in cancer development and response to therapy.
- Record: found
- Abstract: found
- Article: not found