- Record: found
- Abstract: found
- Article: found
Viral Oncology: Molecular Biology and Pathogenesis
Read this article at
Abstract
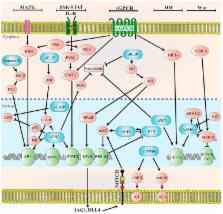
Oncoviruses are implicated in approximately 12% of all human cancers. A large number of the world’s population harbors at least one of these oncoviruses, but only a small proportion of these individuals go on to develop cancer. The interplay between host and viral factors is a complex process that works together to create a microenvironment conducive to oncogenesis. In this review, the molecular biology and oncogenic pathways of established human oncoviruses will be discussed. Currently, there are seven recognized human oncoviruses, which include Epstein-Barr Virus (EBV), Human Papillomavirus (HPV), Hepatitis B and C viruses (HBV and HCV), Human T-cell lymphotropic virus-1 (HTLV-1), Human Herpesvirus-8 (HHV-8), and Merkel Cell Polyomavirus (MCPyV). Available and emerging therapies for these oncoviruses will be mentioned.
Related collections
Most cited references356
- Record: found
- Abstract: found
- Article: not found
Human Papillomavirus Types in Head and Neck Squamous Cell Carcinomas Worldwide: A Systematic Review
- Record: found
- Abstract: found
- Article: not found
Human papillomavirus and cervical cancer.
- Record: found
- Abstract: found
- Article: not found