- Record: found
- Abstract: found
- Article: found
Protein structure alignment beyond spatial proximity
Read this article at
Abstract
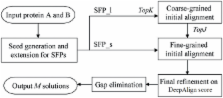
Protein structure alignment is a fundamental problem in computational structure biology. Many programs have been developed for automatic protein structure alignment, but most of them align two protein structures purely based upon geometric similarity without considering evolutionary and functional relationship. As such, these programs may generate structure alignments which are not very biologically meaningful from the evolutionary perspective. This paper presents a novel method DeepAlign for automatic pairwise protein structure alignment. DeepAlign aligns two protein structures using not only spatial proximity of equivalent residues (after rigid-body superposition), but also evolutionary relationship and hydrogen-bonding similarity. Experimental results show that DeepAlign can generate structure alignments much more consistent with manually-curated alignments than other automatic tools especially when proteins under consideration are remote homologs. These results imply that in addition to geometric similarity, evolutionary information and hydrogen-bonding similarity are essential to aligning two protein structures.
Related collections
Most cited references25
- Record: found
- Abstract: found
- Article: not found
Protein homology detection by HMM-HMM comparison.
- Record: found
- Abstract: found
- Article: not found
Protein structure alignment by incremental combinatorial extension (CE) of the optimal path.
- Record: found
- Abstract: found
- Article: not found