- Record: found
- Abstract: found
- Article: found
SETD2: from chromatin modifier to multipronged regulator of the genome and beyond
Read this article at
Abstract
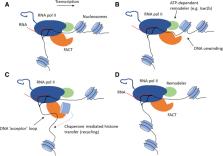
Histone modifying enzymes play critical roles in many key cellular processes and are appealing proteins for targeting by small molecules in disease. However, while the functions of histone modifying enzymes are often linked to epigenetic regulation of the genome, an emerging theme is that these enzymes often also act by non-catalytic and/or non-epigenetic mechanisms. SETD2 (Set2 in yeast) is best known for associating with the transcription machinery and methylating histone H3 on lysine 36 (H3K36) during transcription. This well-characterized molecular function of SETD2 plays a role in fine-tuning transcription, maintaining chromatin integrity, and mRNA processing. Here we give an overview of the various molecular functions and mechanisms of regulation of H3K36 methylation by Set2/SETD2. These fundamental insights are important to understand SETD2’s role in disease, most notably in cancer in which SETD2 is frequently inactivated. SETD2 also methylates non-histone substrates such as α-tubulin which may promote genome stability and contribute to the tumor-suppressor function of SETD2. Thus, to understand its role in disease, it is important to understand and dissect the multiple roles of SETD2 within the cell. In this review we discuss how histone methylation by Set2/SETD2 has led the way in connecting histone modifications in active regions of the genome to chromatin functions and how SETD2 is leading the way to showing that we also have to look beyond histones to truly understand the physiological role of an ‘epigenetic’ writer enzyme in normal cells and in disease.
Related collections
Most cited references222

- Record: found
- Abstract: found
- Article: found
Highly accurate protein structure prediction with AlphaFold
- Record: found
- Abstract: found
- Article: found
Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing
- Record: found
- Abstract: found
- Article: not found