- Record: found
- Abstract: found
- Article: not found
An efficient multi-locus mixed model approach for genome-wide association studies in structured populations
Read this article at

Abstract
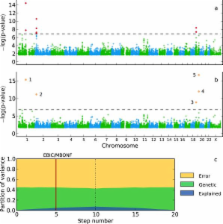
Population structure causes genome-wide linkage disequilibrium between unlinked loci, leading to statistical confounding in genome-wide association studies. Mixed models have been shown to handle the confounding effects of a diffuse background of large numbers of loci of small effect well, but do not always account for loci of larger effect. Here we propose a multi-locus mixed model as a general method for mapping complex traits in structured populations. Simulations suggest that our method outperforms existing methods, in terms of power as well as false discovery rate. We apply our method to human and Arabidopsis thaliana data, identifying novel associations in known candidates as well as evidence for allelic heterogeneity. We also demonstrate how a priori knowledge from an A. thaliana linkage mapping study can be integrated into our method using a Bayesian approach. Our implementation is computationally efficient, making the analysis of large datasets (n > 10000) practicable.
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: not found
Association mapping in structured populations.
- Record: found
- Abstract: found
- Article: not found
Precision mapping of quantitative trait loci.
- Record: found
- Abstract: found
- Article: not found