- Record: found
- Abstract: found
- Article: found
Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18

Read this article at
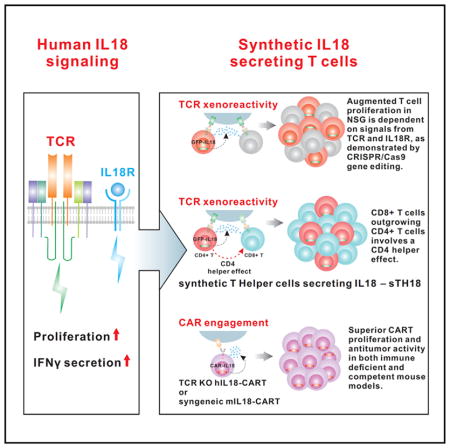
SUMMARY
The effects of transgenically encoded human and mouse IL-18 on T cell proliferation and its application in boosting chimeric antigen receptor (CAR) T cells are presented. Robust enhancement of proliferation of IL-18-secreting human T cells occurred in a xenograft model, and this was dependent on TCR and IL-18R signaling. IL-18 augmented IFN-γ secretion and proliferation of T cells activated by the endogenous TCR. TCR-deficient, human IL-18-expressing CD19 CAR T cells exhibited enhanced proliferation and antitumor activity in the xenograft model. Antigen-propelled activation of cytokine helper ensemble (APACHE) CAR T cells displayed inducible expression of IL-18 and enhanced antitumor immunity. In an intact mouse tumor model, CD19-IL-18 CAR T cells induced deeper B cell aplasia, significantly enhanced CAR T cell proliferation, and effectively augmented antitumor effects in mice with B16F10 melanoma. These findings point to a strategy to develop universal CAR T cells for patients with solid tumors.
In Brief
Hu et al. create IL-18-secreting chimeric antigen receptor T (IL-18-CAR T) cells to significantly boost CAR T cell proliferation and antitumor activity.
Related collections
Most cited references16
- Record: found
- Abstract: found
- Article: not found
Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy.
- Record: found
- Abstract: found
- Article: not found