- Record: found
- Abstract: found
- Article: found
IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment
Read this article at
Abstract
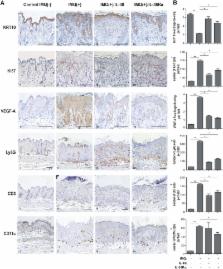
IL-36 cytokines, a subgroup of IL-1 family, comprise IL-36α, IL-36β, and IL-36γ agonists, abundantly expressed in psoriatic skin, and IL-36RA and IL-38 antagonists. In psoriatic skin, IL-36 cytokines interfere with keratinocyte cornification programs and induce the release of antimicrobial peptides and chemokines active on neutrophils and Th17 lymphocytes. To date, the role of IL-38 antagonist in psoriasis remains to be defined. Here, we demonstrate that skin and circulating IL-38 levels are reduced in psoriatic patients and in other skin diseases characterized by neutrophilic infiltrate. In psoriasis, the balance of IL-36γ agonist/IL-38 antagonist serum levels is in favor of agonists and is closely associated with disease severity. Interestingly, IL-38 is upregulated by anti-IL-17A biological treatment and positively correlates with the therapeutic efficacy of secukinumab in psoriatic patients. The downregulation of IL-38 expression is strictly related to keratinocyte de-differentiation triggered by the inflammatory cytokines IL-36γ, IL-17, and IL-22. Finally, we demonstrate that administration of recombinant full-length IL-38 counteracts in vitro the biological processes induced by IL-36γ in human keratinocytes and endothelial cells and attenuates in vivo the severity of the psoriasiform phenotype induced by IMQ in mice. Such effects are achieved by restoring the physiological programs of keratinocyte proliferation and differentiation, and reducing the immune cell infiltrates.
Related collections
Most cited references42
- Record: found
- Abstract: found
- Article: not found
Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis.
- Record: found
- Abstract: found
- Article: not found
Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis.
- Record: found
- Abstract: found
- Article: not found