- Record: found
- Abstract: found
- Article: found
Improvement of genome assembly completeness and identification of novel full-length protein-coding genes by RNA-seq in the giant panda genome
Read this article at
Abstract
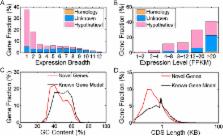
High-quality and complete gene models are the basis of whole genome analyses. The giant panda ( Ailuropoda melanoleuca) genome was the first genome sequenced on the basis of solely short reads, but the genome annotation had lacked the support of transcriptomic evidence. In this study, we applied RNA-seq to globally improve the genome assembly completeness and to detect novel expressed transcripts in 12 tissues from giant pandas, by using a transcriptome reconstruction strategy that combined reference-based and de novo methods. Several aspects of genome assembly completeness in the transcribed regions were effectively improved by the de novo assembled transcripts, including genome scaffolding, the detection of small-size assembly errors, the extension of scaffold/contig boundaries, and gap closure. Through expression and homology validation, we detected three groups of novel full-length protein-coding genes. A total of 12.62% of the novel protein-coding genes were validated by proteomic data. GO annotation analysis showed that some of the novel protein-coding genes were involved in pigmentation, anatomical structure formation and reproduction, which might be related to the development and evolution of the black-white pelage, pseudo-thumb and delayed embryonic implantation of giant pandas. The updated genome annotation will help further giant panda studies from both structural and functional perspectives.
Related collections
Most cited references22

- Record: found
- Abstract: found
- Article: found
Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources
- Record: found
- Abstract: found
- Article: not found
Next-generation transcriptome assembly.
- Record: found
- Abstract: found
- Article: not found