- Record: found
- Abstract: found
- Article: found
Jalview Version 2—a multiple sequence alignment editor and analysis workbench
Read this article at
Abstract
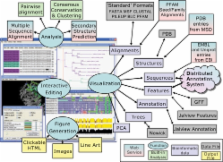
Summary: Jalview Version 2 is a system for interactive WYSIWYG editing, analysis and annotation of multiple sequence alignments. Core features include keyboard and mouse-based editing, multiple views and alignment overviews, and linked structure display with Jmol. Jalview 2 is available in two forms: a lightweight Java applet for use in web applications, and a powerful desktop application that employs web services for sequence alignment, secondary structure prediction and the retrieval of alignments, sequences, annotation and structures from public databases and any DAS 1.53 compliant sequence or annotation server.
Availability: The Jalview 2 Desktop application and JalviewLite applet are made freely available under the GPL, and can be downloaded from www.jalview.org
Contact: g.j.barton@dundee.ac.uk
Related collections
Most cited references24
- Record: found
- Abstract: found
- Article: not found
SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny.
- Record: found
- Abstract: found
- Article: not found
The Distributed Annotation System
- Record: found
- Abstract: found
- Article: not found