- Record: found
- Abstract: found
- Article: not found
Sodium channelopathies of skeletal muscle result from gain or loss of function
Read this article at

Abstract
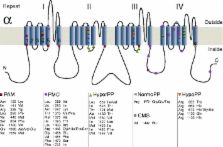
Five hereditary sodium channelopathies of skeletal muscle have been identified. Prominent symptoms are either myotonia or weakness caused by an increase or decrease of muscle fiber excitability. The voltage-gated sodium channel Na V1.4, initiator of the muscle action potential, is mutated in all five disorders. Pathogenetically, both loss and gain of function mutations have been described, the latter being the more frequent mechanism and involving not just the ion-conducting pore, but aberrant pores as well. The type of channel malfunction is decisive for therapy which consists either of exerting a direct effect on the sodium channel, i.e., by blocking the pore, or of restoring skeletal muscle membrane potential to reduce the fraction of inactivated channels.
Related collections
Most cited references61
- Record: found
- Abstract: found
- Article: not found
A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation.
- Record: found
- Abstract: found
- Article: not found
Gating pore current in an inherited ion channelopathy.
- Record: found
- Abstract: found
- Article: not found