- Record: found
- Abstract: found
- Article: not found
Conserved Role of Intragenic DNA Methylation in Regulating Alternative Promoters
Read this article at

Abstract
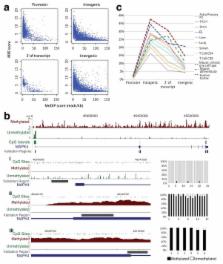
While the methylation of DNA in 5′ promoters suppresses gene expression, the role of DNA methylation in gene bodies is unclear 1– 5 . In mammals, tissue- and cell type-specific methylation is present in a small percentage of 5′ CpG island (CGI) promoters, while a far greater proportion occurs across gene bodies, coinciding with highly conserved sequences 5– 10 . Tissue-specific intragenic methylation might reduce, 3 or, paradoxically, enhance transcription elongation efficiency 1, 2, 4, 5 . Capped analysis of gene expression (CAGE) experiments also indicate that transcription commonly initiates within and between genes 11– 15 . To investigate the role of intragenic methylation, we generated a map of DNA methylation from human brain encompassing 24.7 million of the 28 million CpG sites. From the dense, high-resolution coverage of CpG islands, the majority of methylated CpG islands were revealed to be in intragenic and intergenic regions, while less than 3% of CpG islands in 5′ promoters were methylated. The CpG islands in all three locations overlapped with RNA markers of transcription initiation, and unmethylated CpG islands also overlapped significantly with trimethylation of H3K4, a histone modification enriched at promoters 16 . The general and CpG-island-specific patterns of methylation are conserved in mouse tissues. An in-depth investigation of the human SHANK3 locus 17, 18 and its mouse homologue demonstrated that this tissue-specific DNA methylation regulates intragenic promoter activity in vitro and in vivo. These methylation-regulated, alternative transcripts are expressed in a tissue and cell type-specific manner, and are expressed differentially within a single cell type from distinct brain regions. These results support a major role for intragenic methylation in regulating cell context-specific alternative promoters in gene bodies.
Related collections
Most cited references20
- Record: found
- Abstract: found
- Article: not found
The transcriptional landscape of the mammalian genome.
- Record: found
- Abstract: found
- Article: not found
Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning.
- Record: found
- Abstract: found
- Article: not found