- Record: found
- Abstract: found
- Article: found
EPI-SF: essential protein identification in protein interaction networks using sequence features
Read this article at
Abstract
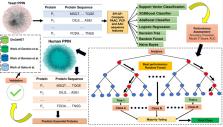
Proteins are considered indispensable for facilitating an organism’s viability, reproductive capabilities, and other fundamental physiological functions. Conventional biological assays are characterized by prolonged duration, extensive labor requirements, and financial expenses in order to identify essential proteins. Therefore, it is widely accepted that employing computational methods is the most expeditious and effective approach to successfully discerning essential proteins. Despite being a popular choice in machine learning (ML) applications, the deep learning (DL) method is not suggested for this specific research work based on sequence features due to the restricted availability of high-quality training sets of positive and negative samples. However, some DL works on limited availability of data are also executed at recent times which will be our future scope of work. Conventional ML techniques are thus utilized in this work due to their superior performance compared to DL methodologies. In consideration of the aforementioned, a technique called EPI-SF is proposed here, which employs ML to identify essential proteins within the protein-protein interaction network (PPIN). The protein sequence is the primary determinant of protein structure and function. So, initially, relevant protein sequence features are extracted from the proteins within the PPIN. These features are subsequently utilized as input for various machine learning models, including XGB Boost Classifier, AdaBoost Classifier, logistic regression (LR), support vector classification (SVM), Decision Tree model (DT), Random Forest model (RF), and Naïve Bayes model (NB). The objective is to detect the essential proteins within the PPIN. The primary investigation conducted on yeast examined the performance of various ML models for yeast PPIN. Among these models, the RF model technique had the highest level of effectiveness, as indicated by its precision, recall, F1-score, and AUC values of 0.703, 0.720, 0.711, and 0.745, respectively. It is also found to be better in performance when compared to the other state-of-arts based on traditional centrality like betweenness centrality (BC), closeness centrality (CC), etc. and deep learning methods as well like DeepEP, as emphasized in the result section. As a result of its favorable performance, EPI-SF is later employed for the prediction of novel essential proteins inside the human PPIN. Due to the tendency of viruses to selectively target essential proteins involved in the transmission of diseases within human PPIN, investigations are conducted to assess the probable involvement of these proteins in COVID-19 and other related severe diseases.
Related collections
Most cited references58
- Record: found
- Abstract: found
- Article: not found
A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug-Repurposing

- Record: found
- Abstract: found
- Article: found