- Record: found
- Abstract: found
- Article: not found
Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus
Read this article at

Abstract
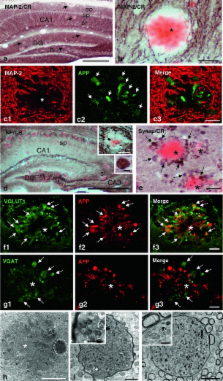
Dystrophic neurites associated with amyloid plaques precede neuronal death and manifest early in Alzheimer’s disease (AD). In this work we have characterized the plaque-associated neuritic pathology in the hippocampus of young (4- to 6-month-old) PS1 M146L/APP 751SL mice model, as the initial degenerative process underlying functional disturbance prior to neuronal loss. Neuritic plaques accounted for almost all fibrillar deposits and an axonal origin of the dystrophies was demonstrated. The early induction of autophagy pathology was evidenced by increased protein levels of the autophagosome marker LC3 that was localized in the axonal dystrophies, and by electron microscopic identification of numerous autophagic vesicles filling and causing the axonal swellings. Early neuritic cytoskeletal defects determined by the presence of phosphorylated tau (AT8-positive) and actin–cofilin rods along with decreased levels of kinesin-1 and dynein motor proteins could be responsible for this extensive vesicle accumulation within dystrophic neurites. Although microsomal Aβ oligomers were identified, the presence of A11-immunopositive Aβ plaques also suggested a direct role of plaque-associated Aβ oligomers in defective axonal transport and disease progression. Most importantly, presynaptic terminals morphologically disrupted by abnormal autophagic vesicle buildup were identified ultrastructurally and further supported by synaptosome isolation. Finally, these early abnormalities in axonal and presynaptic structures might represent the morphological substrate of hippocampal dysfunction preceding synaptic and neuronal loss and could significantly contribute to AD pathology in the preclinical stages.
Related collections
Most cited references38
- Record: found
- Abstract: found
- Article: not found
Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease.
- Record: found
- Abstract: found
- Article: not found
Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment.
- Record: found
- Abstract: found
- Article: not found