- Record: found
- Abstract: found
- Article: found
Genomic evolution and adaptation of arthropod-associated Rickettsia
Read this article at
Abstract
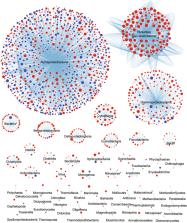
Rickettsia species are endosymbionts hosted by arthropods and are known to cause mild to fatal diseases in humans. Here, we analyse the evolution and diversity of 34 Rickettsia species using a pangenomic meta-analysis (80 genomes/41 plasmids). Phylogenomic trees showed that Rickettsia spp. diverged into two Spotted Fever groups, a Typhus group, a Canadensis group and a Bellii group, and may have inherited their plasmids from an ancestral plasmid that persisted in some strains or may have been lost by others. The results suggested that the ancestors of Rickettsia spp. might have infected Acari and/or Insecta and probably diverged by persisting inside and/or switching hosts. Pangenomic analysis revealed that the Rickettsia genus evolved through a strong interplay between genome degradation/reduction and/or expansion leading to possible distinct adaptive trajectories. The genus mainly shared evolutionary relationships with α-proteobacteria, and also with γ/β/δ-proteobacteria, cytophagia, actinobacteria, cyanobacteria, chlamydiia and viruses, suggesting lateral exchanges of several critical genes. These evolutionary processes have probably been orchestrated by an abundance of mobile genetic elements, especially in the Spotted Fever and Bellii groups. In this study, we provided a global evolutionary genomic view of the intracellular Rickettsia that may help our understanding of their diversity, adaptation and fitness.
Related collections
Most cited references116
- Record: found
- Abstract: found
- Article: not found
Cytoscape: a software environment for integrated models of biomolecular interaction networks.
- Record: found
- Abstract: found
- Article: not found
MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms.
- Record: found
- Abstract: found
- Article: not found