- Record: found
- Abstract: found
- Article: found
Pexophagy is responsible for 65% of cases of peroxisome biogenesis disorders
Read this article at
ABSTRACT
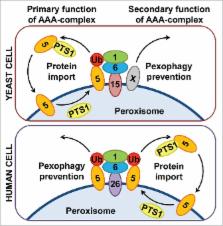
Peroxisome biogenesis disorders (PBDs) is a group of diseases caused by mutations in one of the peroxins, proteins responsible for biogenesis of the peroxisomes. In recent years, it became clear that many peroxins (e.g., PEX3 and PEX14) play additional roles in peroxisome homeostasis (such as promoting autophagic degradation of peroxisomes or pexophagy), which are often opposite to their originally established functions in peroxisome formation and maintenance. Even more interesting, the peroxins that make up the peroxisomal AAA ATPase complex (AAA-complex) in yeast (Pex1, Pex6 and Pex15) or mammals (PEX1, PEX6, PEX26) are responsible for the downregulation of pexophagy. Moreover, this might be even their primary role in human: to prevent pexophagy by removing from the peroxisomal membrane the ubiquitinated peroxisomal matrix protein import receptor, Ub-PEX5, which is also a signal for the Ub-binding pexophagy receptor, NBR1. Remarkably, the peroxisomes rescued from pexophagy by autophagic inhibitors in PEX1 G843D (the most common PBD mutation) cells are able to import matrix proteins and improve their biochemical function suggesting that the AAA-complex per se is not essential for the protein import function in human. This paradigm-shifting discovery published in the current issue of Autophagy has raised hope for up to 65% of all PBD patients with various deficiencies in the AAA-complex. Recognizing PEX1, PEX6 and PEX26 as pexophagy suppressors will allow treating these patients with a new range of tools designed to target mammalian pexophagy.
Related collections
Most cited references24
- Record: found
- Abstract: found
- Article: not found
Genetics and molecular basis of human peroxisome biogenesis disorders.
- Record: found
- Abstract: found
- Article: not found
Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines.

- Record: found
- Abstract: found
- Article: found