- Record: found
- Abstract: found
- Article: found
Mutant Huntingtin induces activation of the Bcl-2/adenovirus E1B 19-kDa interacting protein (BNip3)
Read this article at
Abstract
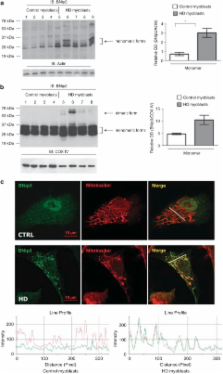
Huntington's disease (HD) is a neurodegenerative disorder characterized by progressive neuronal death in the basal ganglia and cortex. Although increasing evidence supports a pivotal role of mitochondrial dysfunction in the death of patients' neurons, the molecular bases for mitochondrial impairment have not been elucidated. We provide the first evidence of an abnormal activation of the Bcl-2/adenovirus E1B 19-kDa interacting protein 3 (BNip3) in cells expressing mutant Huntingtin. In this study, we show an abnormal accumulation and dimerization of BNip3 in the mitochondria extracted from human HD muscle cells, HD model cell cultures and brain tissues from HD model mice. Importantly, we have shown that blocking BNip3 expression and dimerization restores normal mitochondrial potential in human HD muscle cells. Our data shed light on the molecular mechanisms underlying mitochondrial dysfunction in HD and point to BNip3 as a new potential target for neuroprotective therapy in HD.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease.
- Record: found
- Abstract: found
- Article: not found
Mitochondrial defect in Huntington's disease caudate nucleus.
- Record: found
- Abstract: found
- Article: not found