- Record: found
- Abstract: found
- Article: not found
Microarray-based DNA methylation profiling: technology and applications
Read this article at

Abstract
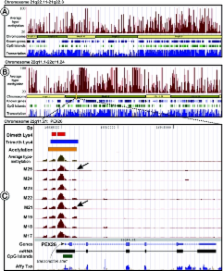
This work is dedicated to the development of a technology for unbiased, high-throughput DNA methylation profiling of large genomic regions. In this method, unmethylated and methylated DNA fractions are enriched using a series of treatments with methylation sensitive restriction enzymes, and interrogated on microarrays. We have investigated various aspects of the technology including its replicability, informativeness, sensitivity and optimal PCR conditions using microarrays containing oligonucleotides representing 100 kb of genomic DNA derived from the chromosome 22 COMT region in addition to 12 192 element CpG island microarrays. Several new aspects of methylation profiling are provided, including the parallel identification of confounding effects of DNA sequence variation, the description of the principles of microarray design for epigenomic studies and the optimal choice of methylation sensitive restriction enzymes. We also demonstrate the advantages of using the unmethylated DNA fraction versus the methylated one, which substantially improve the chances of detecting DNA methylation differences. We applied this methodology for fine-mapping of methylation patterns of chromosomes 21 and 22 in eight individuals using tiling microarrays consisting of over 340 000 oligonucleotide probe pairs. The principles developed in this work will help to make epigenetic profiling of the entire human genome a routine procedure.
Related collections
Most cited references38
- Record: found
- Abstract: found
- Article: not found
Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells.
- Record: found
- Abstract: found
- Article: not found
A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands.
- Record: found
- Abstract: not found
- Article: not found