- Record: found
- Abstract: found
- Article: found
Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma
Read this article at
Abstract
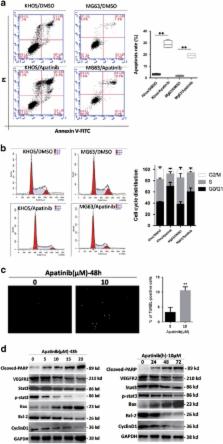
The cure rate of osteosarcoma has not improved in the past 30 years. The search for new treatments and drugs is urgently needed. Apatinib is a high selectivity inhibitor of vascular endothelial growth factor receptor-2 (VEGFR2) tyrosine kinase, exerting promising antitumoral effect in various tumors. The antitumor effect of Apatinib in human osteosarcoma has never been reported. We investigated the effects of Apatinib in osteosarcoma in vitro and in vivo. Osteosarcoma patients with high levels of VEGFR2 have poor prognosis. Apatinib can inhibit cell growth of osteosarcoma cells. In addition to cycle arrest and apoptosis, Apatinib induces autophagy. Interestingly, inhibition of autophagy increased Apatinib-induced apoptosis in osteosarcoma cells. Immunoprecipitation confirmed direct binding between VEGFR2 and signal transducer and activator of transcription 3 (STAT3). Downregulation of VEGFR2 by siRNA resulted in STAT3 inhibition in KHOS cells. VEGFR2 and STAT3 are inhibited by Apatinib in KHOS cells, and STAT3 act downstream of VEGFR2. STAT3 and BCL-2 were downregulated by Apatinib. STAT3 knockdown by siRNA reinforced autophagy and apoptosis induced by Apatinib. BCL-2 inhibits autophagy and was apoptosis restrained by Apatinib too. Overexpression of BCL-2 decreased Apatinib-induced apoptosis and autophagy. Apatinib repressed the expression of STAT3 and BCL-2 and suppressed the growth of osteosarcoma in vivo. To sum up, deactivation of VEGFR2/STAT3/BCL-2 signal pathway leads to Apatinib-induced growth inhibition of osteosarcoma.
Related collections
Most cited references33
- Record: found
- Abstract: found
- Article: not found
Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis.
- Record: found
- Abstract: found
- Article: not found
