- Record: found
- Abstract: found
- Article: not found
Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms
Read this article at

Abstract
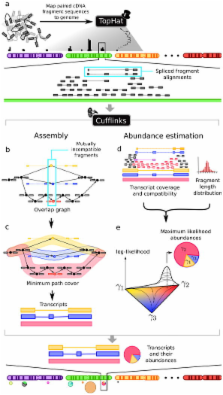
High-throughput mRNA sequencing (RNA-Seq) holds the promise of simultaneous transcript discovery and abundance estimation 1- 3 . We introduce an algorithm for transcript assembly coupled with a statistical model for RNA-Seq experiments that produces estimates of abundances. Our algorithms are implemented in an open source software program called Cufflinks. To test Cufflinks, we sequenced and analyzed more than 430 million paired 75bp RNA-Seq reads from a mouse myoblast cell line representing a differentiation time series. We detected 13,692 known transcripts and 3,724 previously unannotated ones, 62% of which are supported by independent expression data or by homologous genes in other species. Analysis of transcript expression over the time series revealed complete switches in the dominant transcription start site (TSS) or splice-isoform in 330 genes, along with more subtle shifts in a further 1,304 genes. These dynamics suggest substantial regulatory flexibility and complexity in this well-studied model of muscle development.
Related collections
Most cited references17
- Record: found
- Abstract: found
- Article: not found
The transcriptional landscape of the yeast genome defined by RNA sequencing.
- Record: found
- Abstract: found
- Article: not found
miR-145 and miR-143 Regulate Smooth Muscle Cell Fate Decisions
- Record: found
- Abstract: found
- Article: not found