- Record: found
- Abstract: found
- Article: found
KRDS: a web server for evaluating drug resistance mutations in kinases by molecular docking
Read this article at
Abstract
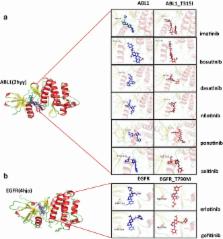
Kinases are major targets of anti-cancer therapies owing to their importance in signaling processes that regulate cell growth and proliferation. However, drug resistance has emerged as a major obstacle to cancer therapy. Resistance to drugs has various underlying mechanisms, including the acquisition of mutations at drug binding sites and the resulting reduction in drug binding affinity. Therefore, the identification of mutations that are relevant to drug resistance may be useful to overcome this issue. We hypothesized that these mutations can be identified by combining recent advances in computational methods for protein structure modeling and ligand docking simulation. Hence, we developed a web-based tool named the Kinase Resistance Docking System (KRDS) that enables the assessment of the effects of mutations on kinase-ligand interactions. KRDS receives a list of mutations in kinases, generates structural models of the mutants, performs docking simulations, and reports the results to users. The changes in docking scores and docking conformations can be analyzed to infer the effects of mutations on drug binding and drug resistance. We expect our tool to improve our understanding of drug binding mechanisms and facilitate the development of effective new drugs to overcome resistance related to kinases; it may be particularly useful for biomedical researchers who are not familiar with computational environments. Our tool is available at http://bcbl.kaist.ac.kr/KRDS/.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
The protein kinase complement of the human genome.
- Record: found
- Abstract: found
- Article: not found
The comprehensive antibiotic resistance database.
- Record: found
- Abstract: found
- Article: not found