- Record: found
- Abstract: found
- Article: not found
Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC
Read this article at

Abstract
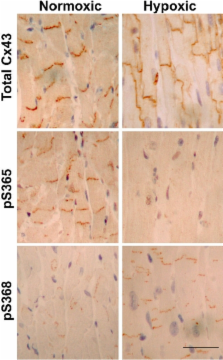
Phosphorylation at unspecified sites is known to regulate the life cycle (assembly, gating, and turnover) of the gap junction protein, Cx43. In this paper, we show that Cx43 is phosphorylated on S365 in cultured cells and heart tissue. Nuclear magnetic resonance structural studies of the C-terminal region of Cx43 with an S365D mutation indicate that it forms a different stable conformation than unphosphorylated wild-type Cx43. Immunolabeling with an antibody specific for Cx43 phosphorylated at S365 shows staining on gap junction structures in heart tissue that is lost upon hypoxia when Cx43 is no longer specifically localized to the intercalated disk. Efficient phosphorylation at S368, an important Cx43 channel regulatory event that increases during ischemia or PKC activation, depends on S365 being unphosphorylated. Thus, phosphorylation at S365 can serve a “gatekeeper” function that may represent a mechanism to protect cells from ischemia and phorbol ester-induced down-regulation of channel conductance.
Related collections
Most cited references38
- Record: found
- Abstract: found
- Article: not found
RADIOAUTOGRAPHIC STUDIES OF CHOLINE INCORPORATION INTO PERIPHERAL NERVE MYELIN
- Record: found
- Abstract: found
- Article: not found
Connexin 26 mutations in hereditary non-syndromic sensorineural deafness.
- Record: found
- Abstract: found
- Article: not found