- Record: found
- Abstract: found
- Article: found
Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy?
research-article
17 August 2017
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Overview
DNA damaging chemotherapy is the first line of treatment for certain cancers, but
its long-term success is often marred by the eventual acquisition of chemoresistance.
Other cancers cannot be treated because they are intrinsically resistant to such chemotherapy.
These 2 types of resistance are coupled in the context of translesion synthesis (TLS),
which is carried out by specialized TLS DNA polymerases that can replicate past DNA
lesions but in a lower fidelity manner. First, TLS DNA polymerases permit the bypass
of modified DNA bases during DNA synthesis, thereby allowing proliferation to continue
in the presence of chemotherapy, an issue of particular relevance to intrinsic drug
resistance. Second, mistakes introduced by TLS polymerases copying over DNA lesions
introduced during the chemotherapy lead to mutations that contribute to acquired resistance.
These dual functions of mutagenic TLS polymerases with respect to chemoresistance
make these proteins very promising targets for adjuvant therapy. The major branch
of mutagenic TLS requires REV1, a Y family DNA polymerase that recruits other TLS
polymerases with its C-terminal domain (CTD) including POL ζ, which is also required.
Recent evidence obtained using mouse models is summarized, which shows that interfering
with REV1/POL ζ-dependent mutagenic TLS during DNA damaging chemotherapy can help
overcome problems due to both intrinsic resistance and acquired resistance. Ways to
develop drugs that block mutagenic TLS are also considered, including taking advantage
of structural knowledge to target key protein-protein interfaces.
Introduction
While DNA damaging chemotherapy can be very effective and even curative in the treatment
of certain cancers, intrinsic and acquired drug resistance underlies tumor progression
and morbidity in many cancer patients. Intrinsic resistance defines a cell state that
is inherently tolerant of drug action. This can include the activation of drug efflux
pumps or detoxifying processes that effectively reduce intracellular drug concentration
[1]. This can also include a change in the recognition or persistence of DNA damage,
mediated by an enhanced DNA repair capability, a blunted DNA damage response, or the
ability to proliferate in the presence of DNA damage. Conversely, acquired drug resistance
represents a mutational or epigenetic process by which a chemosensitive cell develops
1 or more of the characteristics of an intrinsically resistant cancer cell. Thus,
the mechanisms underlying intrinsic and acquired drug resistance are quite distinct.
One describes a cell state, and the other describes the capability of reaching that
cell state. Yet, these processes are very much coupled in the context of mutagenic
translesion synthesis (TLS).
As discussed throughout this review, mutagenic TLS polymerases underlie 2 important
phenotypes in response to genotoxic chemotherapy. First, they allow for the bypass
of modified DNA bases during DNA synthesis, allowing proliferation to continue in
the presence of chemotherapy. Second, the low fidelity replication performed by TLS
polymerases results in the introduction of inappropriate, nonpairing bases across
from modified nucleotides. The bypass function of TLS polymerases is particularly
relevant to intrinsic drug resistance. Many tumors, including most pancreatic adenocarcinomas,
nonsmall cell lung cancers, and aggressive brain tumors, as well as most metastatic
malignancies, fail to significantly regress following chemotherapy [2]. In these tumors,
TLS activity contributes to a drug resistant state by promoting the tolerance of DNA
damage [3–6]. Conversely, the mutational role of TLS polymerases is central to process
of acquired drug resistance. Tumor regression and relapse following chemotherapy is
almost always accompanied by the development of drug resistant disease. This may not
occur at initial relapse, but upon serial cycles of treatment patients generally succumb
to tumors that have acquired intrinsically resistant disease. In fact, for certain
cancers the overall prognosis is not dictated by the initial response of the tumor
to chemotherapy. Rather, the response of the relapsed tumor to therapy is a significantly
better determinant of overall survival. For instance, a high error-prone TLS activity
translates into greater tumor adaptation to chemotherapy, while a low error-prone
TLS activity leaves tumor in a treatment-naïve state. This latter state is amenable
to continued long-term treatment of tumors that remain response to treatment with
the initial therapy.
The dual functions of mutagenic TLS polymerases in intrinsic and acquired chemoresistance
make these proteins very attractive potential targets for adjuvant therapy. When combined
with front-line genotoxic therapy, these TLS inhibitors would be expected to sensitize
tumors to chemotherapy while blocking drug-induced mutation. Consequently, while the
generation of such inhibitors is complex, their route to the clinic is more apparent.
TLS inhibitors could be applied in combination with the standard of care for many
malignancies. By effectively increasing the effects of chemotherapy in target cells,
these agents may also allow for a reduction in chemotherapy dose regimens. An added
benefit of these agents may be a reduction in the rate of secondary chemotherapy-driven
malignancies that occur in patients following successful treatment of the primary
disease.
TLS polymerases bypass DNA damage
TLS polymerases are highly conserved, specialized DNA polymerases that can replicate
past aberrant DNA lesions but in a lower fidelity manner—a trade-off that preserves
genomic integrity in cells [7]. These incorrect nucleotides become fixed into mutations
during the next round of DNA replication, contributing to overall fitness and evolution
in single cell organisms but propelling tumorigenesis and disease in humans (Fig 1A).
There are 10 known human TLS polymerases (REV1, POL η, POL ι, POL κ, POL ζ, POL μ,
POL λ, POL β, POL ν, and POL θ), which are distributed in 4 families (Y, B, X, and
A), and also Prim Pol, which additionally has primase activity. Although all TLS DNA
polymerases are more error-prone than replicative DNA polymerases, some are capable
of bypassing specific (cognate) lesions in a relatively error-free manner (Table 1).
The extent of DNA synthesis errors during TLS depends on various factors, including
the identities of the TLS polymerases employed, the presence or absence of cognate
lesions, DNA sequence context, and thermodynamic favorability in the catalytic step
[8–10]. The significance of the TLS process to human health is illustrated by xeroderma
pigmentosum-variant patients, who are deficient in POL η and are therefore susceptible
to UV radiation-induced cancers because the cognate UV-induced cyclobutane pyrimidine
dimers are instead bypassed by alternate TLS polymerases (POL ι and POL κ) in a relatively
error-prone manner [11, 12].
10.1371/journal.pgen.1006842.g001
Fig 1
DNA damage bypass process.
(A) Mechanism of the 2-step DNA damage bypass process. To bypass DNA damage, REV1
inserts deoxycytidine triphosphates across the damage or orchestrates the recruitment
of the other polymerases, POL ι, POL κ, POL η, to replicate across the damage. Thereafter,
POL ζ complex can help extend beyond the damage to enable re-initiation of undamaged
DNA replication. If an incorrect nucleotide gets incorporated across the damage, this
misincorporated nucleotide will lead to a mutation in the next round of replication.
(B) A schematic representing the protein domains of the Y-family translesion synthesis
(TLS) polymerases, REV1, POL ι, POL κ, POL η.
10.1371/journal.pgen.1006842.t001
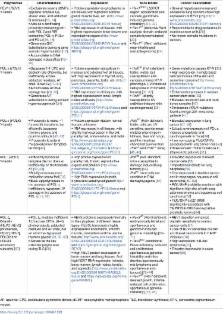
Table 1
Summary of the characteristics, expression, the availability of mouse model, and association
to cancers of B- and Y-family translesion synthesis polymerases.
Polymerase
Characteristics
Expression
Mice Model
Cancer Association
REV1 (REV1)
Y-family
• Exclusively inserts dCMPs opposite template Gs, abasic sites, and adducted G residues
[13, 14]• Acts as a scaffolding protein by interacting with both POL ζ and RIR containing
POL η, POL κ and POL ι [15, 16]• Generates G/C substitutions during Ig gene somatic
hypermutation [17]• Accumulates in DNA damaged induced foci [18–20]
• Protein expression is cytoplasmic in all tissues, with highest in adrenal gland,
muscle, liver, etc. (http://www.proteinatlas.org/ENSG00000135945-REV1/tissue)• RNA
expressed in all tissues, with highest expression in brain tissues and reproductive
organs (http://www.proteinatlas.org/ENSG00000135945-REV1/tissue and https://gtexportal.org/home/gene/REV1)
• Rev1
BRCT (ΔBRCT region; accelerated skin cancers, genotoxin-induced genome instability)
[21, 22].• Rev1
AA (defective Rev1 catalytic domain; reduced somatic hypermutation) [23].• Rev1
KO (Rev1 deficient; near-infertile and unstable genome) [24]
• Several hepatocarcinomas and occasional lung cancers show high expression of REV1
[25] (http://www.proteinatlas.org/ENSG00000135945-REV1/cancer) • Responsible for drug
resistance in ovarian cancer cells [26]• No known somatic mutations in cancers
POL η (POLH)Y-family
• Bypasses T-T CPD and cisplatin-GG efficiently, but inefficiently across adducted
residues, AP sites, 8-oxo-G [27–32]• Accumulates at DNA damage foci [20, 33].• Generates
A/T substitutions during somatic hypermutagenesis [34]
• Protein expression ubiquitous in nucleus and cytoplasm of all tissues, with high
expression in thyroid, lung, pancreas, placenta, testis, etc. (http://www.proteinatlas.org/ENSG00000170734-POLH/tissue)
• RNA expressed in all tissues, with highest expression in tonsil, lymph nodes and
testis (http://www.proteinatlas.org/ENSG00000170734-POLH/tissue and https://gtexportal.org/home/gene/POLH)
• Polh
KO (Pol η deficient; fertile, viable, but susceptible to skin cancers, mirrors XP-V
phenotype, UV irradiated cells prone to chromatid breaks) [35–37]• Polh
+/- (slightly susceptible to UV radiation-induced skin carcinogenesis) [35]
• Gene mutations causes XP-V [38]• High expression in single basal cell carcinomas
of the skin and some liver cancers (http://www.proteinatlas.org/ENSG00000170734-POLH/cancer)
• Enhanced expression in ovarian cancer stem cells [39]• Elevated levels in head and
neck tumor samples [40]• 3 missense POLH mutations found amongst 201 melanoma patients
[41]
POL κ (POLK)Y-family
• Propensity to make −1 frameshift mutations, but efficiently bypasses thymine glycols
and guanine adducts [42, 43]• Propensity to extend mispaired primer-template termini
[44]
• Protein expression data in normal tissues unknown• RNA expressed in all tissues,
with slightly high expression in thyroid, parathyroid, endometrium, and testis (http://www.proteinatlas.org/ENSG00000122008-POLK/tissue#gene_information
& https://gtexportal.org/home/gene/POLK)
Polk
KO (Pol κ deficient; fertile, cells are UV sensitive, spontaneous mutator phenotype
in kidneys, liver and lungs, and the mice has shortened survival than Polk
+/- and Polk
+/+ mice) [45, 46]
• Elevated expression in lung cancer [47, 48]• Ectopic overexpression of POL κ induces
aneuploidy and carcinogenesis in mice [49]• Two non-coding POLK SNPs associated with
lung cancer risk [50]• Three somatic POLK mutations in 26 prostrate patients [51]
POL ι (POLI)Y-family
• Efficiently bypasses template dA; but does so inefficiently on the template dT [52,
53]• Briefly accumulates in replication stress foci [54]• Back-up polymerase in the
absence of POL η. Inefficiently bypasses UV damage in the absence of POL η [11, 55]
• High protein expression in parathyroid, thyroid, reproductive organs and pituitary
(http://www.proteinatlas.org/ENSG00000101751-POLI/tissue) • High RNA expression in
testis, thyroid and parathyroid gland (http://www.proteinatlas.org/ENSG00000101751-POLI/tissue
and https://gtexportal.org/home/gene/POLI)
Poli
KO (Pol ι deficient; mice susceptible to damage-induced lung tumors) [56].
Polι
KO mice cells not sensitive to DNA damaging agents [57]
• Elevated expression in breast cancer cells [58]• Important candidate for lung neoplasia
[59]• Overexpressed in bladder cancer and in esophageal squamous cell carcinoma [60–62]•
POLI SNP (rs8305) correlated with significant high risk of both lung adenocarcinoma
and squamous cell carcinoma [63]• POLI SNP (rs3218786) significantly associated with
TMPRSS2-ERG fusion-positive prostrate tumors [64]
POL ζ4
B-family(REV3 [REV3] polymerase, REV7 [REV7], POLD2 and POLD3 accessory subunits)
[65]
• POL ζ4 mediate inefficient TLS across CPDs, (6–4) photoproducts, adducted residues
and AP sites, but an error free bypass of thymine glycols [53, 66, 67]• Serves as
the key extender polymerase during TLS [68]
• REV3 protein is expressed minimally in the cytoplasm of different tissue types.
REV3L transcript is highly expressed in endometrin, smooth muscle, cerebellum and
the uterine tissues (http://www.proteinatlas.org/ENSG00000009413-REV3L/tissue and
https://gtexportal.org/home/gene/REV3) • High REV7 protein expression in bone marrow
and lung tissues. And high REV7 RNA expression in testis, bone marrow, lymph nodes,
tonsils, and appendix (http://www.proteinatlas.org/ENSG00000116670-MAD2L2/tissue and
https://gtexportal.org/home/gene/MAD2L)
• Rev3
KO (Rev3 deficient; embryonically lethal and spontaneous and genotoxin induced genome
instability) [69–71]• Rev3
Δlox (conditional Rev3 deficiency; reduced cell proliferation, spontaneous genomic
instability and mice develop spontaneously mic lymphoma and spontaneous skin tumors)
[72–74]• Rev7
KO (Rev7 deficient; delayed growth, infertile, reduced cell proliferation, spontaneous
genome instability) [75, 76]
• REV7 depletion enhances cisplatin sensitivity in ovarian cancer cells [77]• Loss
of REV7 sensitizes ovarian and breast cancer cells to PARP inhibition [78]• High expression
in B-cell lymphoma [79]• Elevated expression in colon cancer [80]
AP, apurinic; CPD, cyclobutane pyrimidine dimers; dCMP, deoxycytidine monophosphate;
TLS, translesion synthesis; XP-V, xeroderma pigmentosum-variant.
Distinct structural and biochemical features of the TLS polymerases enable them to
replicate past the DNA damage. For example, in contrast to classical replicative polymerases,
Y-family TLS polymerases possess a smaller thumb and finger domain that makes fewer
contacts with DNA and also lack an 3ʹ-5ʹ exonuclease activity to proofread misincorporated
nucleotides. Together, these structural attributes result in a larger and/or more
permissive catalytic site than replicative polymerases that allows TLS polymerases
to accommodate distorted and damaged nucleotides [81, 82]. In addition, other physical
features such as the polymerase-associated domain of Y-family polymerases and the
wrist and the N-clasp region of POL κ also contribute to polymerase architecture conducive
to replication across DNA damage (Fig 1B) [83–87]. Furthermore, regulatory domains
of TLS polymerases enable their proper localization and regulation [88]. These special
structural features of TLS polymerases are fundamental to their roles in DNA damage
bypass.
Besides the structural features of individual TLS polymerases, successful TLS also
depends on interactions between these polymerases and other cellular proteins that
target and choreograph their activity. REV1 functions as a principle scaffolding protein,
which recruits other TLS polymerases to first insert a nucleotide opposite the DNA
lesion and then eventually help extend the distorted primer-template terminus, in
what is recognized as the two-step mechanism of TLS (Fig 2) [7, 8, 89]. For the insertion
step, a particular interface of the REV1 CTD interacts with the REV1-interacting-region
(RIR) of the inserter polymerases (POL η, POL ι, POL κ). Mutations that disrupt the
RIR-interface in the Rev1 CTD prevent interaction with the inserter polymerase in
yeast-2 hybrid (Y2H) screens [15, 16, 90, 91]. Insertion across from the damaged base
can also be less frequently carried out by REV1 and POL ζ [8]. In the second step,
an extender TLS enzyme, a role most frequently fulfilled by POL ζ (REV3/REV7/POLD2/POLD3)
and in some cases by POL κ, replaces the inserter and extends the primer-template
termini [90]. For the POL ζ -mediated extension step, a different interface in REV1
CTD—distinct from the interface for RIR recognition—makes contact with specific amino
acids located on REV7. Mutating residues in the Rev7-interface of the Rev1 CTD inhibits
Rev1-Rev7 interaction in Y2H studies and sensitizes chicken DT40 cells to cisplatin
[15]. Apart from bypassing DNA damage at stalled replication forks, TLS polymerases
also engage in filling single stranded (ss) DNA gaps left behind by replicative polymerases,
via the less-well understood gap-filling mechanism [92, 93].
10.1371/journal.pgen.1006842.g002
Fig 2
Protein-protein interactions between translesion synthesis (TLS) polymerases are important
for the DNA damage bypass process.
Two pathways are expected to facilitate TLS across DNA damage—the REV1 dependent and
REV1 independent pathway. Majority of the DNA lesions are bypassed in a REV1 dependent
fashion, which engages in protein-protein interactions with other TLS polymerases
via its C-terminus. REV1 interacts with the REV1-interacting-region (RIR)-containing
residues of POL ι, POL κ, POL η to enable insertion of nucleotides across the damage.
And REV1 also interacts via key residues with REV7 of the POL ζ complex to facilitate
extension beyond the insertion step. REV1 also binds to POLD3 subunit of the POL ζ
complex to enable the key switch from the “insertion” to the “extension” step. In
the REV1 independent pathway, the RIR-containing polymerases, POL ι, POL κ, POL η,
by interacting with the proliferating cell nuclear antigen (PCNA) interacting protein
(PIP) and ubiquitin-binding motif (UBM)/ ubiquitin-binding zinc finger (UBZ) domains
of PCNA, can also enable TLS at the damaged site. Likewise, the POL ζ complex also
interacts with the PIP box of PCNA to access the DNA and enable TLS.
Interestingly, TLS polymerases are also required for other cellular functions. For
example, during interstrand cross-link (ICL) repair in replicating cells, certain
TLS polymerases—REV1, POL ι, POL κ and POL ν—are required for DNA synthesis over the
ICL on the newly exposed leading strand [94–97]. Likewise, in nonreplicating cells,
ICL repair depends on the Rev1-POL ζ TLS polymerases to fill the ssDNA-gaps [98].
In a similar fashion, both nucleotide excision repair (NER) and base excision repair
(BER) pathways respectively can employ POL κ and POL η to fill the ssDNA gaps left
behind after the excising step [99, 100]. Additionally, POL η was recently shown to
drive microhomology-mediated break-induced replication (MMBIR) that causes complex
genomic rearrangements in yeast and has an important role in homologous recombination
(HR) in DT40 cells [101, 102]. Finally, REV1 was recently shown to be required for
replication of G-quadruplex structures, thereby influencing epigenetic stability [103].
Independent of its role in TLS, REV7 promotes nonhomologous end joining (NHEJ) at
double strand breaks and at telomeres by inhibiting CtIP-mediated end resection [104].
Additionally, REV7 plays a supporting role in cell cycle regulation by sequestering
CDH1, which prevents premature activation of the anaphase-promoting complex, thereby
inhibiting an exit from mitosis [105]. All these examples are suggestive of an overarching
influence of TLS polymerases and their components on cellular physiology, in which
they influence DNA damage tolerance, DNA repair, epigenetic stability, and replication
across repetitive sequences.
Modulation of TLS polymerases alters tumor response to chemotherapy
A growing body of evidence now shows that suppression of TLS polymerases not only
sensitizes tumor cells to drugs, but also reduces acquisition of drug-induced mutations
implicated in tumor resistance. Thus, inhibition of TLS polymerases is a promising
new approach to improving cancer therapy. Moreover, in some cancers, TLS polymerases
are overexpressed (Table 1),
The impact TLS polymerases have on chemotherapy responses in different cancer subtypes
has recently been investigated. In one study, the potential of Rev3 inhibition for
the treatment of intrinsically chemoresistant cancers was investigated. A study utilizing
the Kras
G12D
;p53
−/−
preclinical model of lung adenocarcinoma showed that, when the level of Rev3 was reduced,
these otherwise resistant tumors were sensitized to cisplatin, increasing the overall
survival of mice with Rev3-deficient tumors by 2-fold compared with control mice with
Rev3-proficient tumors [106]. Reduction of Rev3 or Rev1 in these tumor cells also
reduced cisplatin-induced mutagenesis in culture.
In a study that employed the Eμ-myc arf
-/- mouse model of B-cell lymphoma, when mice were subjected to repeated cycles of
tumor engraftment and cyclophosphamide treatment, relapsed tumors that appeared after
the first round of chemotherapy continued to respond to cyclophosphamide if they were
Rev1 deficient. This is in direct contrast to Rev1-proficient relapsed tumors, which
exhibited varying degrees of acquired resistance to cyclophosphamide chemotherapy
(Fig 3). Additionally, cyclophosphamide-induced mutagenesis of these lymphoma cells
in culture was suppressed by Rev1 depletion. These studies showed that Rev1-dependent
error-prone bypass of cyclophosphamide-induced DNA damage contributes to the mutagenesis
and hence the tumor drug resistance. Thus this study provided the first in vivo evidence
that TLS polymerases play a critical role in the development of acquired chemoresistance
[107].
10.1371/journal.pgen.1006842.g003
Fig 3
Reduction of Rev1 suppresses chemoresistance.
In a tumor mouse model, administration of chemotherapy reduces tumor formation by
killing the generally chemoresensitive tumor cells. However, many of the tumors that
relapse are resistant to further killing from chemotherapeutic treatment, thereby
reducing survival of the mice. In contrast, mice harboring relapsed tumors in which
REV1 has been knocked down remain sensitive to chemotherapy, whereby their survival
is prolonged.
Chemotherapy-induced mutagenesis is a phenomenon proposed to cause secondary malignancies
and tumor relapse. Hence, targeting REV1 and REV3 might not only increase killing
of cancer cells but could also potentially suppress secondary malignancies and tumor
relapse. The same principal was explored when an innovative nanoparticle-mediated
delivery system was used to target both REV1 and REV3 in combination with a cisplatin
prodrug. A nearly complete inhibition of tumor growth and dramatically enhanced survival
was observed in LnCaP prostate cancer mouse model [108]. In addition, REV7 depletion
has been shown to sensitize ovarian cancer to cisplatin and reduce tumor volumes in
nude mice [77]. These studies support the hypothesis that TLS inhibition can suppress
at least some classes of intrinsic chemoresistance. Likewise depletion of REV3 in
cervical cancer cells [109] or nonsmall cell lung cancer cells [110]; REV1, POL ζ,
POL η in HeLa cells [111]; and POL η in ovarian cancer stem cells [39] all sensitize
cells to cisplatin. It remains to be seen whether other cancer cell subtypes would
similarly respond to knockdown of TLS polymerases and whether observations in cell
studies could be recapitulated in mouse models.
Another approach to potentially enhance tumor cell killing via suppression of TLS
polymerases is to discover synthetic lethal partners of TLS polymerases. For example,
this classical approach is employed in killing BRCA2-deficient tumors by utilizing
PARP1 inhibitors [112]. Although a compelling idea, TLS synthetic partners are largely
unknown. However, a whole genome siRNA library screen in A549 lung cancer cells identified
one gene RRMI—the large subunit of ribonucleotide reductase that confers a synthetic
lethal interaction with REV3 [113]. In another lung cancer cell line and in breast
cancer cells, ataxia-telangiectasia and Rad3 related inhibition was found to synthetically
enhance lethality in cisplatin-treated REV3-deficient cells [114]. In addition, Rev3-deficient
DT40 cells exhibited synthetic lethality with RAD54 [115], suggesting a promising
potential. Synthetic-lethal partners of TLS polymerases need to be explored in greater
detail across other cancer subtypes.
Drug inhibitors to target TLS polymerases
Taken together, the studies discussed above suggest that small molecules that directly
inhibit catalytic functions or disrupt key protein-protein interactions of TLS polymerases
could be adjuvants that have the potential to significantly improve chemotherapy.
For example, fluorescence-based assays conducted in high-throughput platforms were
used to search for small molecule inhibitors that affect catalytic functions of TLS
polymerases. Pamoic acid, aurintricarboxylic acid, and ellagic acid were found to
inhibit POL ι and POL η [112], while candesartan cilexetil inhibited the enzymatic
function of POL κ as well as enhanced UV-induced cytotoxicity in xeroderma pigmentosum-variant
(XP-V) cells [116]. Likewise, 3-O-methylfunicone, a natural compound isolated from
a marine fungal strain, selectively inhibited mammalian Y-family TLS polymerase activity
(POL κ, POL, ι, POL η) [117]. Further studies are required to identify compounds with
improved specificity and potency.
Very recently small molecules inhibitors that target TLS DNA polymerase protein-protein
interactions have been shown to be possible therapeutic candidates. For example, a
small molecule inhibitor that binds to REV7 and inhibits its interaction with REV3
was shown to partially suppress ICL repair [118]. Whether the same drug could also
suppress TLS is worth investigating. Similarly, detailed structural knowledge of other
TLS interfaces, such as between REV1 and REV7 and between REV1 and RIR carrying proteins
could be exploited in drug discovery and design.
Perspective and conclusion
Inhibiting TLS polymerases is a promising approach to improve chemotherapy as it could
increase killing of cancer cells, while at the same time reducing the possibility
of relapse and acquired drug resistance by reducing chemotherapy-induced mutagenesis.
Even cancers known to be intrinsically drug resistant could potentially be sensitized
by this approach. Additionally, TLS specific inhibition could also potentially target
other repair and recombination pathways that involve TLS polymerases including NER,
BER, MMBIR, HR, and NHEJ. However, several outstanding questions still need to be
addressed, for example, improving understanding of the structural basis of key protein-protein
interactions made by the TLS polymerases. Recently it was shown that the subunits
of replicative polymerases cross talk with TLS Polymerases. For instance, the POLD3
subunit of the replicative DNA polymerase POL δ possess an RIR that interacts with
the RIR-interface of REV1 CTD, while the POLD2 subunit of POL δ interacts with POL
η [90]. These observations suggest that the TLS mechanism is even more complex than
previously anticipated and that drug inhibitors for 1 TLS polymerase could potentially
target multiple other TLS polymerases. An added complication is that TLS polymerases
η, ι, and κ can also function independently of REV1 by interacting with proliferating
cell nuclear antigen (PCNA) via the UBM/UBZ domain and the PCNA interacting protein
(PIP) domain (Fig 2). It is not known quantitatively what percent of DNA damage in
the cells is bypassed in a Rev1-dependent versus REV1-independent manner. This knowledge
will help decipher whether a single inhibitor targeting the Rev1/RIR or the REV1/REV7
interaction or a combination of inhibitors targeting the REV1/RIR, REV1/Rev7 and UBM/UBZ-PIP-PCNA
interactions would be required for a complete TLS inhibition. Also, a better understanding
of synthetic lethal partners of TLS polymerases would provide insights into which
tumors might be most susceptible to chemotherapy treatments involving small molecule
inhibitors of TLS polymerases. Finally, the effectiveness of small molecule inhibitors
of TLS polymerase could be further improved by delivery systems that could target
these drugs to specific tumors in cancer patients. Because protein-protein interactions
are so important for TLS, drug targets for these interaction interfaces could be promising
candidates for cancer therapeutics.
Related collections
Most cited references109
- Record: found
- Abstract: found
- Article: not found
Stromal biology and therapy in pancreatic cancer.
Albrecht Neesse, Patrick Michl, Kristopher K Frese … (2011)
- Record: found
- Abstract: found
- Article: not found
The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta.
C Masutani, R Kusumoto, A. Yamada … (1999)
- Record: found
- Abstract: found
- Article: not found
Mechanism of replication-coupled DNA interstrand crosslink repair.
M Räschle, Orlando D. Schärer, Jack Griffith … (2008)