- Record: found
- Abstract: found
- Article: found
Functional DNA demethylation is accompanied by chromatin accessibility
Read this article at
Abstract
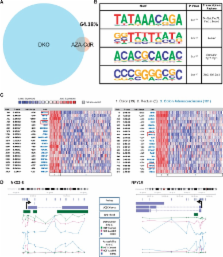
DNA methylation inhibitors such as 5-aza-2′-deoxycytidine (5-Aza-CdR) are currently used for the treatment of myelodysplastic syndrome. Although global DNA demethylation has been observed after treatment, it is unclear to what extent demethylation induces changes in nucleosome occupancy, a key determinant of gene expression. We use the colorectal cancer cell line HCT116 as a model to address this question and determine that <2% of regions demethylated by 5-Aza-CdR treatment assume an open configuration. Consolidating our findings, we detect nucleosome retention at sites of global DNA methylation loss in DKO1, an HCT116-derived non-tumorigenic cell-line engineered for DNA methyltransferase disruption. Notably, regions that are open in both HCT116 cells after treatment and in DKO1 cells include promoters belonging to tumor suppressors and genes under-expressed in colorectal cancers. Our results indicate that only a minority of demethylated promoters are associated with nucleosome remodeling, and these could potentially be the epigenetic drivers causing the loss of tumorigenicity. Furthermore, we show that the chromatin opening induced by the histone deacetylase inhibitor suberoylanilide hydroxamic acid has strikingly distinct targets compared with those of 5-Aza-CdR, providing a mechanistic explanation for the importance of combinatorial therapy in eliciting maximal de-repression of the cancer epigenome.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
Cancer genetics and epigenetics: two sides of the same coin?
- Record: found
- Abstract: found
- Article: not found
Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer.
- Record: found
- Abstract: found
- Article: not found