
- Record: found
- Abstract: found
- Article: found
Chromosome-Level Assembly of Artemia franciscana Sheds Light on Sex Chromosome Differentiation
Read this article at
Abstract
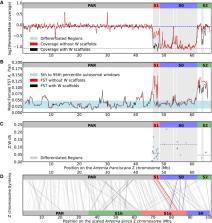
Since the commercialization of brine shrimp (genus Artemia) in the 1950s, this lineage, and in particular the model species Artemia franciscana, has been the subject of extensive research. However, our understanding of the genetic mechanisms underlying various aspects of their reproductive biology, including sex determination, is still lacking. This is partly due to the scarcity of genomic resources for Artemia species and crustaceans in general. Here, we present a chromosome-level genome assembly of A. franciscana (Kellogg 1906), from the Great Salt Lake, United States. The genome is 1 GB, and the majority of the genome (81%) is scaffolded into 21 linkage groups using a previously published high-density linkage map. We performed coverage and F ST analyses using male and female genomic and transcriptomic reads to quantify the extent of differentiation between the Z and W chromosomes. Additionally, we quantified the expression levels in male and female heads and gonads and found further evidence for dosage compensation in this species.
Related collections
Most cited references58

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.
- Record: found
- Abstract: found
- Article: not found